Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Technology Feature
- Published: 11 January 2022

Method of the Year: protein structure prediction
- Vivien Marx 1
Nature Methods volume 19 , pages 5–10 ( 2022 ) Cite this article
38k Accesses
33 Citations
41 Altmetric
Metrics details
- Machine learning
- Protein folding
- Structure determination
Nature Methods has named protein structure prediction the Method of the Year 2021.
You have full access to this article via your institution.
If the Earth moves for you, among other reasons, the causes can be geologic or romantic. In science, in the context of predicting protein structure, you might have felt the ground tremble in late 2020 as you perused the results of the 14th Critical Assessment of Protein Structure Prediction (CASP). In this competition, scientists regularly test the prowess of their methods that computationally predict the intricate twirly-curly three-dimensional (3D) structure of a protein from a sequence of amino acids.
A pleasant frisson may have set in more recently as you browsed the new and rapidly growing AlphaFold Protein Structure Database or perused papers 1 , 2 , 3 about a method called AlphaFold and its application to the entire human proteome, or when you dug into the code that drives this inference engine, with its neural network architecture that yields the 3D structure of proteins from a given amino acid sequence. The team behind AlphaFold is DeepMind Technologies, launched as an AI startup in 2010 by Demis Hassabis, Shane Legg and Mustafa Suleyman and now part of Alphabet after being acquired by Google in 2014. DeepMind has presented AlphaFold1 4 and AlphaFold2 and, more recently, AlphaFold-Multimer 5 for predicting the structures of known protein complexes.
AlphaFold has received much attention, but there are many other recent tools from academic labs, such as RoseTTAFold 6 , a method with a ‘three-track’ network architecture developed in the lab of David Baker and colleagues at the University of Washington along with academic teams around the world. It can be used to, for example, predict protein structures and generate models of protein-protein complexes, too. In their paper, the authors note that they had been “intrigued” by the DeepMind results and sought to increase the accuracy of protein structure prediction as they worked on their architecture.
At CASP14 in 2020, AlphaFold2 blew away its competitors. The difference between the DeepMind team results and those of the group in second place “was a bit of a shock,” says University College London researcher David Jones. “I’m still processing that a bit, really.” Only some months later, when DeepMind gave a glimpse of its method and shared the code, were scientists able to begin looking under the hood. No new information was used to transition AlphaFold1 to AlphaFold2; there was no “clever trick,” says Jones. The team used what academics had been doing for years but applied it in a more principled way, he says.
In the lead-up to the 2018 CASP13 competition, which the DeepMind team won quite handily with AlphaFold1, Jones had consulted for DeepMind. Especially after machine-learning-based methods were introduced in 2016, CASP results had been steadily improving, says Dame Janet Thornton, DBE, from the European Bioinformatics Institute (EBI). Thornton is the former EBI director and has long worked on the challenges of protein structure determination. She was interviewed jointly with Jones. At CASP13, she had been delighted to see progress taking place with protein structure prediction methods. Now, as Thornton considers the possibilities AlphaFold2 opens up, by having solved a big methods puzzle in science, “it gives me a spring in my step.” She says she hadn’t thought “we’d get quite this far in my lifetime.”
Historical build
The way AlphaFold2 can predict a protein structure is the culmination of a scientific journey that began with the work 7 , 8 of Max Perutz and John Kendrew of the Cavendish Laboratory at the University of Cambridge, says Aled Edwards of the University of Toronto and the Structural Genomics Consortium, a public–private venture. Perutz and Kendrew received the Nobel Prize in 1962 for the way they used X-rays passing through crystallized protein and onto film to painstakingly decipher the structures of proteins such as hemoglobin and myoglobin.
Structural biologists have since followed in their footsteps to experimentally determine structures of many proteins. The research community has deposited structures and accompanying data in the Protein Data Bank (PDB) 9 , an open resource founded in 1971 that holds 185,541 structures as Nature Methods goes to press.
The PDB’s holdings stem from labs around the world that toiled with X-ray crystallography, nuclear magnetic resonance spectroscopy (NMR) or electron microscopy to determine the complex structure of a 3D protein. AlphaFold2’s machine-learning algorithm was trained on the PDB’s data to assess the patterns with which amino acids become the many combinations of helices, sheets and folds that enable a protein to do its specific tasks in a cell.
Converting experimental signals into structures has been the realm of physicists and mathematicians who devoted time, perseverance and sweat to determine protein structures, says Edwards. In the early days, this work involved assessing measurements on photographic film. The fact that they, and those who followed, have been so committed to data quality enabled the continued work in protein structure determination. Speaking more generally, he says, experimentally solving protein structures is “a pain in the (expletive).” It’s why he applauded the foresight of University of Maryland researcher John Moult, who launched CASP in 1994 to highlight and advance community activity related to methods for computationally predicting protein structure. Edwards and many others were part of the NIH-funded Protein Structure Initiative that ran from 2000 to 2015. The project set out to systematically add to PDB’s experimentally determined structures and has certainly contributed to AlphaFold’s success, says Edwards. When the project’s funding ceased, many labs were dismayed. The PSI had been sampling the still-unexplored “structure space,” he says. After the PSI ended, the PDB kept growing as labs continued to add their structures.
The PDB’s main database has been reserved for structures resolved experimentally and by single methods such as X-ray crystallography, NMR or cryo-electron microscopy (cryo-EM), says Helen Berman, who co-founded the Protein Data Bank. Over time, computational models emerged that used multiple sequence alignments and, later, also machine learning to predict structures. PDB-Dev was set up as a digital home for structures determined with “integrative methods,” which means it’s for structures generated using experimental methods combined with computational ones. The strictly in silico structures are held in the ModelArchive .
“AlphaFold is a triumph,” says Berman. But it “would never ever have succeeded, ever,” she says, if models had been improperly mixed with experimentally determined structures. The training set for AlphaFold’s neural network has been PDB’s well-curated experimental data. DeepMind, in collaboration with EBI, is now filling the AlphaFold Protein Structure Database with hundreds of thousands of computationally generated human protein structures and those from many other organisms, including the ‘classic’ research organisms maize, yeast, rat, mouse, fruit fly and zebrafish.
Every day, the PDB sees around 2.5 million downloads of protein coordinates, says Berman. Biotech and pharma companies regularly download the database for research performed behind their firewalls. Around the time of CASP13 in 2018, Berman noticed massive downloads that seemed unlike the typical downloads from the structural biology community. Usage is not monitored in detail, and all of it, be it from academia or companies, has made her happy about the resource. “If you don’t have people use it, then why have it?” she says. As a child of the 1960s, her personal commitment has been to the “public good” that the resource provides. Over time, the PDB team has navigated expanding its global reach and managing structural data generated by emerging methods. “Now we have to make a new kind of decision,” says Berman, whose workload belies the fact that she recently retired from PDB and her position on the Rutgers University faculty. She has daily calls about how the existing data—the data in the PDB, data generated by AlphaFold2, data generated by RoseTTAFold and other platforms, and other computationally generated data—should be stored and, separately, how they should be served to the community.
Rather than make a centralized behemoth of a database, says Janos Hajdu, who splits his time between the European Extreme Light Infrastructure at the Academy of Sciences of the Czech Republic and the Laboratory of Molecular Biophysics at Uppsala University and who is not involved in these discussions, he would like to see “independent databases that talk to each other.”
The actual database plan is still emerging, and details are under wraps until it’s worked out, says Berman. It will take six months to a year to hammer out these details and find a solution that works for all.
Confidence measures
Each AlphaFold2 structure is accompanied by a “confidence score,” which, as Janet Thornton says, will help and guide users, be they structural biologists or scientists working in other areas. The per-residue confidence score (pLDDT) is between 0 and 100.
Indeed, says Aled Edwards, confidence scores are important pieces of information, but they likely matter more to structural biologists than to other biologists. A diabetes researcher with a hypothesis about a protein who has downloaded a structure with a confidence score of 82% will not be deterred from an experiment he or she is planning, he says. The confidence score could be a point that a reviewer might critically note: the paper authors had chosen to use a “maybe structure,” one with a lower confidence score.
Janos Hajdu sees value in confidence scores. Just as one dresses differently for a weather forecast of a 5% or a 95% probability of rain, confidence scores are important and need to be sufficiently well developed. After all, different parts of a predicted structure can have different quality and reliability. The reliability of interpretation also has a human factor to contend with, says Hajdu: even though a lottery win and a lightning strike of a person walking in a storm have similar probabilities, people generally feel less fearful about lightning strikes and more hopeful about their chances of hitting the jackpot.
Into the machine
Compared to other software developed in the academic community, says Thornton, AlphaFold’s advances include more accurate placement of side chains in the protein models and an improved approach to integrating machine learning with homology modeling, which looks at protein structure in the context of evolutionarily related proteins. The software uses homology modeling at an “ultrafine” level, says Jones. “It’s taking little pieces of everything it needs from the whole of PDB.” Instead of taking an homologous 3D structure, building a model from that and then including the side chains and loops, the system finds all the right pieces in high-dimensional space. In a way, he says, it’s solving “the worst jigsaw puzzle in history made up of tiny little pieces.”
In CASP13, DeepMind entered its AlphaFold1, and then in CASP14 the team entered AlphaFold2. A big difference between CASP13 and CASP14, says Jones, was the way the DeepMind team applied language modeling, specifically the self-attention model, to reduce the need for computing steps run sequentially. The leap made the academic community look like “we’d all been spending 30 years staring at the wall doing nothing,” says Jones, which, of course, is not the case. DeepMind’s computational approach is based on one that Google Brain scientists presented at the 2017 Conference on Neural Information Processing Systems called ‘Attention is all you need’ 10 . It’s had great impact on AlphaFold, bioinformatics and the computer science community, he says.
Applying this approach in AlphaFold pares back the recurrent layers that the encoder–decoder architectures in machine learning apply and replaces them with “multi-headed self-attention,” which interconnects many operations at the same time. These attention models “can just mix data across all the data you feed in,” says Jones. Such data-mixing on a scale larger than previously accomplished, lifted constraints that academic groups had faced. Removing computational constraints gives AlphaFold its power to juggle data. “They can mix it up in any way necessary to solve the problem,” he says. At the time of CASP14, bioinformaticians were not yet applying this technology, but since then, says Jones, in machine-learning circles he encounters many scientists who work on variations of attention models.
‘Attention’ is indeed part of a big change in this field, says Burkhard Rost, a computational biologist at the Technical University of Munich who was previously at Columbia University. Both AlphaFold1 and AlphaFold2 rely on multiple sequence alignment and on machine learning. When combining these techniques, academics have used standard feedforward networks with a network of processing units, or nodes, that are arranged in layers with outputs from one layer leading to the next. Training weights the nodes. By including natural-language processing techniques in AlphaFold and in academic labs such as his and others, researchers have enabled machines to ‘learn’ the grammar of a given protein sequence, says Rost, and the grammar gives context. Based on sentences from Wikipedia, a neural network can extract grammar rules for general language. In the same way, the network can extract the grammar of “a protein language,” he says, one that is learned from input amino acid sequence and the corresponding 3D output.
CASP14 felt like being “hit by a truck or a freight train,” says Burkhard Rost. “I’m utterly impressed by what they did.”
A platform can learn, for example, that the amino acid alanine might be both at position 42 and 81 in a protein. But it’s the 3D environment around these amino acids that affects the protein in different ways. Even though this computational approach does not teach 3D structure or evolutionary constraints, systems can learn rules such as physical constraints that shape protein structure. Rost says that never before has there been a CASP winner from outside the field of protein structure prediction. CASP14 felt like being “hit by a truck or a freight train,” he says. He found AlphaFold1’s predictions to be “amazingly accurate.” AlphaFold2 is “a completely different product” in which he sees “so much novelty” he says. “I’m utterly impressed by what they did.”
To train the system, the DeepMind approach used tensor processing units (TPUs), which are Google’s proprietary processors. They are not for sale; academics can only access them through the Google Cloud . Indeed, DeepMind has “great hardware,” says Juan Restrepo-López, a physicist who has turned to biology as a PhD student in the lab of Jürgen Cox at the Max Planck Institute of Biochemistry. AlphaFold2 is likely inconceivable without that hardware, says Restrepo-López. AlphaFold1, with its convolutional neural networks (CNNs), is “for sure much easier to understand due to its simpler architecture.” Both AlphaFold1 and AlphaFold2 were trained on TPUs. AlphaFold1 could be run on graphics processing units (GPUs), and this has also eventually become true for AlphaFold2, he says. In AlphaFold2, DeepMind no longer used CNNs but rather transformers, says Restrepo-López. The main advantage for AlphaFold2 came from Google’s huge computing clusters, which made it possible to run many types of models. “You can go crazy and run 200,000 experiments because you have unlimited resources,” he says. To generate structures, DeepMind first uses multiple sequence analysis, which originated in academia. The core of the algorithm uses transformers, developed at Google. Transformers originated in the field of natural-language processing and are now being applied in many areas. “They are particularly interesting because they can detect long correlations,” he says.
This AlphaFold2 architecture with transformers makes it possible, as previously mentioned, to process many aspects of the sequence in parallel and figure out long-term dependencies very well,says Restrepo-López. For example, residues far apart in a sequence can be very close in a folded protein, and this concept has to be introduced into a model.

For decades, academic groups around the world have been predicting structures using the millions of amino acid sequences in databases and integrating evolutionary information as part of homology modeling. But DeepMind has used many more sequences plus a different way of scaling computation, says Rost.
When he saw CASP13 results, Konstantin Weissenow, now a PhD student in the Rost lab, was a master’s degree student working on a protein structure prediction method. It seemed to him that DeepMind was taking a “traditional” deep learning approach not unlike his. At the time, DeepMind was not sharing the code, but Weissenow felt he could reverse engineer the method and “this is essentially what I tried to do,” he says. He incorporated what he gleaned into his method. But CASP14 and AlphaFold2 “was a different story.” A few months later, Deep Mind made the AlphaFold2 code public. Michael Heinzinger, another graduate student in the Rost lab, was wrapping up protein language modeling as he watched the livestream of CASP14, which the Rost lab was competing in with Weissenow’s tool. When experimentalists began saying that this computational system was reaching close to the quality of experimentally generated results and structures, Heinzinger felt like it was a moment that “people might actually then read in the history books years or decades after this point,” he says. “This was just mind blowing.”
“The big impact came with CASP14,” says Weissenow. By then he had started his PhD work in the Rost lab. He and others had entered their software tool, called EMBedding-based inter-residue distance predictor (EMBER), for CASP14. It’s geared toward predicting protein structures for which there are few evolutionary relatives, and computationally it uses a many-layered convolutional network similar to that of AlphaFold1. EMBER allows the team to predict structures on a large scale, and it can predict the human proteome on a typical computer. It was not going to be as good as AlphaFold2, says Rost, but it has a lower carbon footprint. After CASP14, says Weissenow, some participants got together to consider reverse engineering AlphaFold2, but they soon realized that was not going to work. Then, DeepMind published predictions for 98.5% of the human proteome 2 . This was a few weeks before Weissenow had planned to present his tool at a conference and show how it could generate structures of the human proteome. “Scooped again,” says Rost, who was interviewed jointly with Heinzinger, Weissenow and postdoctoral fellow Maria Littmann, who works on ways to predict, from amino acid sequence, which residues bind DNA, metal or small molecules.
One issue Littmann faced around 2018 and 2019, says Rost, was the lack of experimental data. It will now be interesting, says Litmann, to see how she and others can integrate the availability of these models into their work and extend it. When predicting residues only from sequence but without a structure, “you don’t know what the actual binding site looks like,” she says. In the folded structure, residues may be close together or far apart, and it’s impossible to know, for example, if two residues are part of the same or a different DNA-binding site. “For that she needs a model,” says Rost. Now, given AlphaFold2, Littmann feels she can move beyond the task of predicting which residues bind to being able to predict binding sites.
“This is a game-changer for several applications we are pursuing in the lab,” says Jürgen Cox.
AlphaFold has immense value for work in his lab, says the MPI’s Cox. He finds AlphaFold2 is enabling for proteomics more generally. His team integrates structure information into the lab’s computational-mass-spectrometry-based proteomics workflows, and Restrepo-López is integrating AlphaFold2 predictions into the Cox lab’s MaxQuant algorithms. AlphaFold has trumped a number of existing tools in the protein prediction space, but many of them had been close to retirement age, says Cox. The best way to predict structural information along the protein sequence such as secondary structure or solvent accessibility “is to just do the 3D structure prediction and project these properties from the structure onto the sequence.” With the advent of AlphaFold2, says Cox, it’s become possible to assume that a structure—either a computationally generated or an experimentally deciphered one—is at researchers’ fingertips for nearly every protein and organism and that a computationally generated structure is similar in quality to one determined by X-ray crystallography. “This is a game-changer for several applications we are pursuing in the lab,” he says.
Science or engineering?
To some, AlphaFold’s achievement is more an engineering feat than a scientific one. AlphaFold2’s utility is indisputable, says Jürgen Cox. Every achievement in the development of algorithms and computational tools runs into the issue of being perceived as ‘just’ engineering as opposed to ‘real’ science,” he says. But it’s not justified in this case or in other aspects of computational biology. “Think of the BLAST algorithm. Is it science or engineering?” he asks. Bioinformatics supports life science research and, in so doing, enables findings not achievable through other means. Advances in machine-learning methods are science unto themselves, he says. Differentiating between science and engineering doesn’t matter, says Hajdu, given that tool-making is an integral part of science. “A drill, an XFEL, various algorithms, mathematical breakthroughs” can all turn into tools in some fields, he says, referring to X-ray free electron lasers. “Someone’s science today is someone’s tool tomorrow.”
“You can’t do the science without the engineering,” says Jones. He is essentially ‘split’ across engineering and science in that he holds a double appointment at University College London: in computer science and in structural and molecular biology. “If the science is wrong, it doesn’t matter how good your engineering is,” he says. And if there is bad engineering, no correct answers are to be had. “Engineering makes things a reality,” he says. “And the science builds the foundations on which that happens.”
When a company does it
Some researchers have been irked that a commercial venture achieved this goal of large-scale protein prediction, as opposed to an academic lab or consortium. “I was just pleased overall,” says Thornton, who feels the achievement will benefit the entire field. “In a way it was quite disappointing,” she says, but it’s a company with access to “a lot of compute” and one positioned at the forefront of machine learning.
To Hajdu, it makes no difference that a company and not an academic group reached this goal, he says. Going forward, scientists now have access to many more protein structures, most of them computationally generated. The situation is comparable to one with the sequencing of the human genome, which both a company and an academic consortium worked on. “The important thing is that it is done,” he says. And it matters that the results and tools are or will be available to all. That, he hopes, is an aspect the research community will be able to shape.
When Littmann first saw the CASP14 results, she assumed that because a company had developed the method, it would be kept “behind closed doors,” which would prevent the academic community from ever figuring out how the team had achieved what they had. She also assumed one would have to pay to obtain structures, meaning that the academic community would still have needed other methods to predict structure. Her eye-opener moment was when the DeepMind team announced that they are publishing for the research community’s benefit the structures from UniProt , which is the database of protein sequences, for the entire human proteome. “That’s something that I never expected,” she says. Gone was the situation of a lack of high-resolution structures for most proteins. Now, she says, researchers can revisit projects done with sequences and see if they can improve them by adding a structure to their analysis.
Isabell Bludau, a postdoctoral fellow and computational biologist in the lab of Matthias Mann at the Max Planck Institute of Biochemistry, picked up on the excitement in the research community about AlphaFold, but its “real impact” on her and for her work, she says, also occurred when DeepMind published structures for the entire human proteome, dramatically expanding the structures available. “This information can now be easily integrated into any systems biology analysis that I do,” says Bludau. As she explores patterns in proteomic data, she can now complement information about the presence and quantity of proteins with structural information, meaning that her analysis can provide a more complete picture. “This is, for me, probably the most exciting part of it,” she says.
Local muscle
AlphaFold was trained on the Protein Data Bank, and the DeepMind team used tensor processing units (TPUs), which are Google’s proprietary processors, to do so. Academics can access them through the Google Cloud . As of the end of 2021, AlphaFold could not only be run locally, any TPU constraint was removed, says Burkhard Rost. There is AlphaFold Colab , with which users can predict protein structures using, as the team indicates, a “slightly simplified version of AlphaFold v2.1.0.” This sets up an AlphaFold2 Jupyter Notebook in Google Colaboratory, which is a proprietary version of Jupyter Notebook hosted by Google that offers access to powerful GPUs. A user can ‘execute’ the Python code from a browser on a local computer. AlphaFold2 will run on Google hardware, which might be CPUs, GPUs or TPUs depending on a researcher’s needs. Separately, researchers have developed a Colab notebook called ColabFold AlphaFold2 for predicting protein structures with AlphaFold2 or RoseTTAFold.
The developers include Martin Steinegger at Seoul National Laboratory, who is one of the co-authors of the AlphaFold2 1 paper, Sergey Ovchinnikov and his team at Harvard University, and colleagues at other institutions. Graduate student Konstantin Schütze is part of the developer team; he’s been a member of the Rost lab and has been working in the Steinegger lab as part of his master’s degree research. As the Rost lab’s Michael Heinzinger explains, ColabFold speeds up AlphaFold2 protein prediction many times over, mainly by accelerating the way multiple sequence alignments are generated with Steinegger’s MMseqs2 , which is software for iterative protein sequence searching. Users can install ColabFold locally by following the tips on Konstantin Schütze’s section of the ColabFold github page . The ability to run AlphaFold2 on GPUs can remove dependency on Google infrastructure, says Heinzinger, because one can choose to install AlphaFold2 on one’s own machine. Multiple sequence alignments can be generated on Steinegger’s servers, he says, “so you do not even have to compute your MSAs locally.”
A landscape of change
AlphaFold is poised to change the structural biology community in a number of ways. The AlphaFold–EBI database gives scientists around the world a “global picture” of the data, says Jones, and this might change the discipline of biology itself.
Early in Jones’s career, when he interacted with biologists, he heard them say that protein structure mattered little to their work. Proteins are, he says as he recalls their words, “just blobs that do things and they stick to other blobs.” As a PhD student in Thornton’s lab, he felt differently about protein structures and began working on computational tools for predicting and analyzing them. Labs these days that use cryo-electron tomography (cryo-ET) and cryo-EM are revealing ever more about the structure of ‘blobs’, says Thornton. Resolution with cryo-ET is improving and can reach 1.2 Å, she says, although it’s still generally “relatively low.” For some biological questions, “a blob is enough,” she says. But she and Jones both believe the computationally generated models can help many labs to assess proteins, for instance by fitting the computational structure onto the ‘blob’ they captured with cryo-ET or cryo-EM experiments. What will change overall because of the wealth of computationally generated structures that are becoming available, says Jones, is that the field of structural biology will need to spend less time on technology and thus have more time for assessing why solving structures matters. It will be possible, he says, to appreciate the power of models and the predicted protein structure coordinates for exploring deeper questions.
As Janet Thornton considers the possibilities AlphaFold2 opens up by having solved a big methods puzzle in science, “it gives me a spring in my step.”
Jones and Thornton have many entries on their to-do list of things they wish to understand: the protein folding pathway, protein–protein and protein–DNA interactions, intrinsically disordered proteins, the interactions of proteins with small molecules, questions of drug design, protein complexes, molecular machines and the overarching question of what proteins do. Having a complete proteome of structures opens entirely new avenues for research questions involving the complexity of protein function. When trying to, for instance, explore and understand protein–protein interactions, it’s “quite difficult if you don’t have protein structures,” says Thornton. “It’s not easy when you have them,” says Jones, and, says Thornton, “it’s impossible when you don’t have them.” They both laughed as they said this.
Among the problems Cox and his team want to tackle is predicting the effect of post-translational modifications on the structure of proteins and complexes. Speaking more generally, Hajdu says, the next chapter of research in this area “has just turned absolutely wonderful.” Not only is there much room to improve the methodology, there are tremendous new opportunities to explore using the new tools. “The scale of possibilities is huge,” he says.
AlphaFold2 does not show, says Thornton, how the path of protein folding occurs, how flexibility shapes protein function or what happens to a structure once it’s stabilized with a ligand. At the moment, machine learning struggles with such problems, she says. AlphaFold cannot predict how a mutation affects a protein such that it folds differently or becomes less stable, an effect that lies at the core of many diseases and disorders. “It hasn’t seen all the variants,” says Jones, so it cannot extrapolate how changes affect a protein’s flexibility or stability. In the wake of AlphaFold, some scientists will likely shift their focus. Thornton has observed that “the crystallographers were the most crushed” by AlphaFold and have privately expressed concern that their skills are no longer needed. In the near future, says Cox, he does not see crystallographers as endangered. “Structural information of whole complex structures still requires experiments,” he says. But “the combination of cryo-EM with AlphaFold2 predictions will pose a threat to crystallographers soon.”
In 1970, Walter Hamilton, a chemist and crystallographer at Brookhaven National Laboratory, published a paper 11 in which he stated that determining a molecular structure by crystallography is routine and that “we have reached the day when such a determination is an essential part of the arsenal of any chemist interested in molecular configuration—and what chemist is not?” Hamilton worked on the molecular and crystal structure of amino acids. “The professional crystallographers really got on his case,” says Berman, for saying it had become routine to experimentally determine the structure of small molecules. They were concerned, she says, that he was putting them out of a job, which didn’t happen. And, says Thornton, it’s not happening now.
AlphaFold is shifting the research landscape, though, says Thornton, given that protein structures will be available for most any amino acid sequence. Over time, X-ray crystallographers have become electron microscopists, she says. “They’re looking at bigger complexes, bigger sets or they’re doing electron tomography.” As such, they are colleagues needed for the next phase in structural biology.
The research community is now in the same place with protein structures as it was with small-molecule structure, says Berman. Back in the day, Berman and her merry band of like-minded junior scientists petitioned Hamilton and others to set up the PDB 7 . “We were very young, we talked a lot, we were so excited about looking at the structures,” she says. Hamilton and others did finally agree, but he unfortunately passed away at age 41.
“Ever since I was a postdoc, I’ve really started to appreciate how enabling cryo-EM was for structural biology,” says Bastian Bräuning, who leads a project group in the lab of Brenda Schulman at the Max Planck Institute of Biochemistry. He completed his PhD research in protein crystallography and dabbled, as he says, in cryo-EM. Now he sees how AlphaFold can help with cryo-ET, which produces lower-confidence data than single-particle cryo-EM, but is leading to ever better predictions for parts of bigger protein complexes. Thus, he says, AlphaFold2 “will really enable cryo-electron tomography, too.” Says Bräuning, “I’ve gone from one revolution to the next between my PhD and my postdoc.” Once the big shock and surprise to structural biologists settles in and “you really start looking at the opportunities it gives to you, it becomes less worrying,” he says. “There’s still so much to be done, and not one method or one revolution is going to solve everything.” To a large extent, he says, to characterize proteins bound to small ligands one still needs crystallographic data, which these days are generated at large synchrotrons. This approach is high throughput and is used to screen ligands in a way that cryo-EM cannot yet deliver.
AlphaFold2 is likely to affect a small subset of researchers in negative ways, in that this platform has leapfrogged over their works in progress, says Edwards. He mainly interacts with structural biologists and, to them, solving a structure enables their thinking about a biological problem and guides the design of their next experiment. Traditionally, he says, the “big paper” in the academic world has gone to the scientists who solved the structure, not the person who explained the science of that structure. But he hopes a shift can now take place such that more emphasis will be placed on creative scientific insights about the functions of structures. The academic literature contains fewer than 10 papers on half of the proteins that the human genome generates, says Edwards. Understanding more proteins and more about function is going to help to understand disease. Having structures enables the “‘what is life?’ question,” he says, and the questions about what these proteins do. “The vastness of what we don’t know is the coolest thing in biology.”
Jumper, J. et al. Nature 596 , 583–589 (2021).
Article CAS Google Scholar
Tunyasuvunakool, K. et al. Nature 596 , 590–596 (2021).
AlQuraishi, M. Nature 596 , 487–488 (2021).
Senior, A. W. et al. Nature 577 , 706–710 (2020).
Evans, R. et al. Preprint at https://doi.org/10.1101/2021.10.04.463034 (2021).
Baek, M. et al. Science https://doi.org/10.1126/science.abj8754 (2021).
Perutz, M. F. et al. Nature 185 , 416–422 (1960).
Kendrew, J. C. et al. Nature 185 , 422–427 (1960).
Anonymous Nat. New Biol. 233 , 223 (1971).
Google Scholar
Vaswani, A. et al. Preprint at https://arxiv.org/pdf/1706.03762.pdf (2017).
Hamilton, W. Science 169 , 133–141 (1970).
Download references
Author information
Authors and affiliations.
Nature Methods http://www.nature.com/nmeth
Vivien Marx
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Vivien Marx .
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Marx, V. Method of the Year: protein structure prediction. Nat Methods 19 , 5–10 (2022). https://doi.org/10.1038/s41592-021-01359-1
Download citation
Published : 11 January 2022
Issue Date : January 2022
DOI : https://doi.org/10.1038/s41592-021-01359-1
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Packaging monoamine neurotransmitters.
- Cornelius Gati
Cell Research (2024)
Can AlphaFold’s breakthrough in protein structure help decode the fundamental principles of adaptive cellular immunity?
- Benjamin McMaster
- Christopher Thorpe
- Hashem Koohy
Nature Methods (2024)
AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms
- Nicola Bordin
- Ian Sillitoe
- Christine Orengo
Communications Biology (2023)
Protein structure prediction from the complementary science perspective
- Jorge A. Vila
Biophysical Reviews (2023)
Rethinking the protein folding problem from a new perspective
European Biophysics Journal (2023)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.


An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Comput Struct Biotechnol J
Deep learning for protein secondary structure prediction: Pre and post-AlphaFold
Dewi pramudi ismi.
a Department of Computer Science and Electronics, Faculty of Mathematics and Natural Sciences, Universitas Gadjah Mada, Yogyakarta, Indonesia
b Department of Infomatics, Faculty of Industrial Technology, Universitas Ahmad Dahlan, Yogyakarta, Indonesia
Reza Pulungan
Graphical abstract.

- • Deep neural networks-based predictors have enhanced the PSSP accuracy.
- • There is still an accuracy gap in PSSP, the PSSP theoretical limit expands to 94%.
- • The use of pre-trained LMs as input has successfully improved PSSP performance.
- • PSSP without using evolutionary information is a potential future work.
This paper aims to provide a comprehensive review of the trends and challenges of deep neural networks for protein secondary structure prediction (PSSP). In recent years, deep neural networks have become the primary method for protein secondary structure prediction. Previous studies showed that deep neural networks had uplifted the accuracy of three-state secondary structure prediction to more than 80%. Favored deep learning methods, such as convolutional neural networks, recurrent neural networks, inception networks, and graph neural networks, have been implemented in protein secondary structure prediction. Methods adapted from natural language processing (NLP) and computer vision are also employed, including attention mechanism, ResNet, and U-shape networks. In the post-AlphaFold era, PSSP studies focus on different objectives, such as enhancing the quality of evolutionary information and exploiting protein language models as the PSSP input. The recent trend to utilize pre-trained language models as input features for secondary structure prediction provides a new direction for PSSP studies. Moreover, the state-of-the-art accuracy achieved by previous PSSP models is still below its theoretical limit. There are still rooms for improvement to be made in the field.
1. Introduction
Proteins play important roles for living organisms due to their diverse functions, for example, acting as a catalyst in cell metabolism, playing an essential role in DNA replication, forming cell structures, forming living tissues, and constructing antibodies for the immune system. Protein has four different levels of structure: primary structure, secondary structure, tertiary structure, and quaternary structure. The protein’s primary structure is the amino acid sequence composing its polypeptide chain. The protein’s secondary structure is a local conformation formed within the polypeptide chain due to the hydrogen bonds between atoms in the backbone. The tertiary structure of protein exposes the three-dimensional structure of the protein in the physical space. The quaternary structure of protein is made up of multiple polypeptide chains that come together.
Proteins function differently from one another due to variations in their structures, mainly due to the folds that make up varying tertiary structures [1] , [2] . Since the function of a particular protein is influenced by its tertiary structure, understanding the protein’s tertiary structure is necessary to reveal its functionality. Hence, protein tertiary structure prediction is a crucial task in structural bioinformatics. Prior to the invention of AlphaFold [3] , predicting protein tertiary structure directly from its primary structure was a challenging problem [4] , [5] , [6] , [7] . Thus, protein structure prediction was divided into subsidiary tasks that are easier to accomplish and beneficial for ultimate protein tertiary structure prediction. Researchers attempted to build models that solve these subsidiary tasks.
Protein secondary structure prediction (PSSP) is one of the subsidiary tasks of protein structure prediction and is regarded as an intermediary step in predicting protein tertiary structure. If protein secondary structure can be determined precisely, it helps to predict various structural properties useful for tertiary structure prediction. For example, secondary structures affect structural folds in the polypeptide chain [8] , [9] , and the same protein secondary structures define similar folds in the protein’s polypeptide chain [10] . PSSP started when Pauling et al. [11] suggested α -helices and β -sheets as the dominant conformations even before the first protein structure was revealed. The first protein structure was revealed using an X-ray by Kendrew et al. [12] . PSSP has gone through five generations since then [13] . Table 1 shows the generations and their respective traits.
The history of PSSP studies.
Leveraging evolutionary information as an input feature and utilizing deep neural networks (DNN)-based architectures has become the primary approach done by PSSP researchers in recent years. Three main reasons that warrant the use of DNN in PSSP are: (1) the increasing number of protein sequences in the Protein Data Bank (PDB), (2) the need to understand and capture long-range interactions in protein sequences, and (3) the capability of DNN to observe the underlying characteristics and hidden patterns in protein sequences.
Considering the immense usage of DNN in PSSP recently, we aim to provide a comprehensive literature review on PSSP studies that leverage DNN. Although there have been several publications on the topic, such as [39] , [40] , [41] , [42] , several key points differentiate this review from previous reviews, namely:
- 1. This review focuses on the implementation and architecture details of DNN methods utilized in PSSP;
- 2. This review discusses various approaches that have been applied to improve PSSP accuracy, such as multi-task learning, ensemble learning, iterative learning, attention mechanism, and language modeling;
- 3. This review discusses the more recent and advanced methods employed in PSSP, such as the involvement of pre-trained language models (LMs) and knowledge distillation; and
- 4. This review discusses the progress and perspectives of PSSP in two different time plots, namely before and after the invention of AlphaFold.
This review is organized as follows: general description, data, PSSP in pre-AlphaFold publication, new approaches for feature generation, AlphaFold and its impacts, PSSP in post-AlphaFold publication, and summary and outlook.
2. General description
PSSP can be seen as a classification problem that categorizes an amino acid residue to the type of secondary structure in which it is located. A typical PSSP model takes a sequence of amino acids as input and returns a sequence of the corresponding secondary structure (see Fig. 1 ). There are two different types of PSSP: three-state PSSP and eight-state PSSP. In three-state PSSP, the secondary structure elements consist of helix (H), sheet (E), and coil (C). Helix and sheet are the two main conformations suggested by Pauling et al. [11] . Coil (C) denotes an amino acid that does not fit both H and E. In eight-state PSSP, proposed by Kabsch and Sander [43] , the secondary structure elements consist of α -helix (H), 3 10 -helix (G), parallel/anti-parallel β -sheet conformation (E), isolated β -bridge (B), bend (S), turn (T), π -helix (I), and coil (C). The three-state PSSP is a coarse-grained classification of secondary structure elements, whereas the eight-state PSSP provides more specific traits of the secondary structure elements. Several transformation rules to map the eight-state secondary structure to the three-state secondary structure are proposed by researchers, including DSSP [43] , DEFINE [44] , and Rost and Sander [45] .

A PSSP model takes sequences of amino acids as an input and produces the sequences of corresponding secondary structure elements.
In 2020, four-state and five-state PSSP alternatives were proposed by Shapovalov et al. [13] . The reason for proposing four-state and five-state PSSP was because isolated β -bridge (B) and bend (S) have a small number of samples and low true-positive rates. In five-state PSSP, B and S are considered as C, whereas in four-state PSSP, B, S, and G are considered as C. Moreover, 75% of π -helix (I) was located at the beginning or the end of an α -helix structure (H), so it was categorized as H.
A PSSP model takes a sequence of amino acids as input. Features representing the sequence of amino acids are generated and used to train the PSSP model. Like other machine learning-based systems, DNN-based PSSP models are developed through the training and evaluation phase. The general framework of DNN-based PSSP is shown in Fig. 2 . Based on the framework depicted in the figure, three aspects play essential roles in producing high prediction accuracy: the training data, the features representing the sequence of amino acids, and the model’s architecture. A typical PSSP study usually tackles one or more aspects.

The general framework of PSSP models: two phases in model development, namely training and evaluation. The training dataset is used to build the model, while the test dataset is used to confirm the performance of the trained model.
A DNN-based PSSP model incorporates one or more deep learning methods in its architecture, such as recurrent neural networks (RNN), including long short-term memory (LSTM) and gated recurrent unit (GRU), convolutional neural networks (CNN), inception networks, and other networks. Although most previous PSSP studies focused on developing novel architectures, several PSSP studies focused on different objectives. Several studies focused on proposing new feature representations, such as new amino acid encoding schemes [46] , [47] , [48] , and other features such as protein language models [49] , [50] . Several studies focused on evaluating the evolutionary information used as input of the PSSP model, for example [51] , [52] , [53] , [54] , [55] . Other studies proposed novel architectures as well as input features, for example [36] , [56] . Besides the model’s architecture and features, several studies also proposed new training and test datasets that eliminate data redundancy such that the models trained using these datasets can achieve a good performance [13] , [57] , [58] .
The performance of PSSP models is usually assessed by predicting the secondary structures of benchmark test datasets. It is measured by the prediction accuracy, namely the percentage of correctly classified amino acid residues in the test datasets. Q3 denotes the accuracy of three-state PSSP, whereas Q8 denotes the accuracy of eight-state PSSP. In addition to accuracy, several PSSP studies also used other standard classification performance metrics, such as precision, recall, and F1 score. PSSP model performance is also measured by segment overlap (SOV). SOV calculates segment-based accuracy that tolerates false prediction at the segment’s boundary but penalizes false prediction in the middle of the segment [59] . In 1999, Zemla et al. [60] proposed SOV’99, which normalizes the SOV score such that the final SOV score is on a percentage scale. SOV is unable to extend allowance when more residues can be predicted correctly in a particular segment. This issue becomes the drawback of SOV and SOV_Refine was proposed by Liu and Wang [61] to tackle the issue. Although less prevalent in PSSP, Kappa Performance was used to measure the performance of the PSSP model proposed by [62] , and Matthew’s correlation coefficient [63] was used in [64] .
3.1. Training data
The development of DNN-based PSSP models requires two different datasets: training and test datasets. There are two different fashions with which the researchers obtain training and test datasets. The first is to collect the datasets from the protein database by themselves. The second is to use the benchmark datasets previously collected by other researchers. Researchers self-collected datasets from the protein data bank (PDB) [65] to build their PSSP models [ [13] , [34] , [35] , [37] , [47] , [53] , [56] , [66] , [67] , [68] , [69] , [70] , [71] , [72] , [73] , [74] , [75] ]. In this case, the size of culled datasets varies depending on the date when the data collecting is performed.
Protein data used to train the DNN-based PSSP models are typically obtained using the PISCES CullPDB server [76] . A sequence identity cutoff of 25–30% usually applies. Besides using the PISCES CullPDB server, protein sequences are also obtained from ASTRAL [77] . Several PSSP studies used datasets extracted from ASTRAL including [78] , [68] , [79] . Proteins from the Genbank database [80] were used as training data to build the PSSP model developed by Xavier and Thirunavukarasu [81] . Li et al. [82] predicted the secondary structure of transmembrane proteins and took the protein sequences from the OPM database [83] .
Instead of performing self data assembly, some researchers prefer to use existing datasets from previous researchers. Several well-known benchmark datasets have been used to train DNN-based PSSP (see Table 2 ).
PSSP training datasets. The dataset name usually indicates the number of data included in the dataset. CullPDB dataset usually refers to the dataset collected by [84] .
AlQuraishi [57] published the ProteinNet dataset, a standardized dataset for training and evaluating PSSP models. It was motivated by the availability of standard datasets in computer vision enabling researchers to build models and evaluate their performances using standardized data. The ProteinNet dataset consists of protein sequences, MSAs, position-specific scoring matrices (PSSMs), and training-validation-test splits. The dataset was designed to support the critical assessment of protein structure prediction (CASP) [87] . AlQuraishi [57] collected all protein sequences available in the protein databases before the date of each CASP challenge commencing. The dataset is designed to provide the complete set of protein sequences needed to build PSSP models to solve the respective CASP challenge. For example, the dataset provides protein sequences and structures available in the databases before the date of the CASP10 challenge to train the PSSP models to be tested on CASP10.
Although there are various benchmark datasets for PSSP model training, previous PSSP researchers tend to use different training datasets to maximize the performance of their PSSP models, i.e., using a combination of several benchmark datasets or culling the training dataset from the protein database by themselves.
3.2. Test data
Although PSSP researchers may take the test data from the same source as the training data by culling a portion of the training data as the test dataset, several independent benchmark test datasets are widely used to compare the performance among PSSP models. CASP datasets from the biennial CASP events [87] are notable benchmark test datasets for PSSP studies. There have been 14 CASP events conducted to date; hence there are 14 CASP datasets publicly available [87] . Besides CASP datasets, several benchmark test datasets used to evaluate PSSP performances are shown in Table 3 .
PSSP benchmark test datasets.
4. PSSP in pre-AlphaFold publication
This section focuses on the DNN-based PSSP models released within five years before the publication of AlphaFold (2016–2021). The discussion covers the features representing amino acid sequences, the architecture of PSSP models, and the performance achieved by those models.
4.1. Features
The features representing amino acid sequences can usually be divided into three types: standard features, single-sequence, and other features.
Standard features. The standard input features representing an amino acid sequence in DNN-based PSSP models are the one-hot encoding of amino acids, sequence profiles, and physicochemical properties of amino acids. A one-hot encoding of amino acids is a matrix of size n × 20 , where n denotes the number of amino acids in the sequence, and 20 denotes the number of types of amino acids. Matrix entries corresponding to the amino acid types are filled with 1, while the remaining entries are filled with 0.
The sequence profile is derived from MSA and represents evolutionary information of the sequence. Two different sequence profiles are commonly used in previous PSSP works: position-specific scoring matrix (PSSM) and hidden Markov model (HMM) profiles. PSSM exhibits the probability of each amino acid residue at each position in the sequence based on the MSA. On the other hand, HMM profile offers state transition probability and emission probability as additional information in the sequence profile. PSSM is usually obtained using PSI-BLAST [92] , while the HMM profile is usually obtained using HHBlits [93] . Besides PSI-BLAST and HHBlits, MMSeq2 [94] , an optimized search for massive datasets, is also used to generate sequence profiles. The details of one-hot encoding of amino acids and PSSM are depicted in Fig. 3 .

One hot encoding of amino acids and position specific scoring matrix (PSSM).
Sequence profiles have been used as fundamental features for PSSP models. A sequence profile is generated based on the known protein sequences in the target dataset, i.e., UniRef90. With the significant growth of the number of known protein sequences in the target dataset, the computational resources and time needed to perform PSSP, especially for generating sequence profiles, also increase. Juan et al. [95] proposed an approach to increase the speed of PSSP without compromising accuracy. The approach was carried out by reducing the size of the target dataset by taking a random sampling and reducing the sequence homology of the target dataset to 25% of the sequence identity. This study showed that reducing the size of the target dataset decreased the time cost of PSSP, whereas reducing the sequence homology of the target dataset improved the complexity of the generated PSSM and enhanced the PSSP accuracy.
Several studies performed critical appraisals on the use of sequence profiles in PSSP. Urban et al. [55] performed a study to critically evaluate the role of sequence profiles in the performance of PSSP models. Sequence profiles have shown the ability to increase the performance of PSSP models. A significant accuracy gap exists between the PSSP models using sequence profiles and those using only amino acid sequences. However, its underlying reason has yet to be clearly explained because only amino acid sequences play a role when protein folds are constructed. Moreover, the study was also based on the observation that although training and test datasets used in the prediction have less than 25% of sequence identity, sequences in training and test datasets are likely to come from the same family [96] , [97] , [98] . In that case, a high profile similarity between training and test datasets occurs, resulting in performance evaluation bias. The prediction accuracy of models utilizing sequence profiles is enhanced by the redundancy found in the sequence profiles of the training and test datasets which lead to an invalid evaluation result. Urban et al. [55] also proposed an evaluation protocol, named EVALPro, to measure the performance of the PSSP models with the assessment of profile similarity between training and test proteins. EVALPro employed Gaussian process regression (GPR) to portray the relationship between the profile similarity of training and test proteins and the accuracy gained.
In addition to the one-hot encoding of amino acids and the sequence profiles, the physicochemical properties of amino acids are commonly used as an input feature for the PSSP model. The physicochemical properties include the steric parameter (graph shape index), polarizability, normalized Van der Waals volume, hydrophobicity, isoelectric point, helix probability, and sheet probability. The values of those parameters usually are those specified in Table 1 of [28] .
Single-sequence prediction. Several studies have begun to develop PSSP models that only accept one-hot encoding of amino acids and do not use sequence profiles. It is motivated by the argument that protein structures are only influenced by the amino acid sequences and not by evolutionary information. The PSSP models in this category include SPIDER3-Single [74] , ProteinUnet [99] , SPOT-1D-Single [100] , and S4PRED [70] . With only using amino acid encoding, the highest reported Q3 accuracy is 75.3%, achieved by S4PRED on the CB513 dataset [70] .
Non-standard features. Besides the standard and sequence-only features, previous PSSP studies also used non-standard features and evaluated their impacts on the performance of the PSSP model. Hanson et al. [36] proposed a predicted contact map [101] as an additional input feature in the PSSP model and showed that using a contact map successfully increases accuracy. A conservation score was proposed as an input feature for eCRRNN [38] . However, the effect of this conservation score on eCRRNN accuracy was not mentioned.
4.2. Model architectures
Various deep learning methods have been employed in the architecture of previous PSSP models. We collected all the PSSP publications, including journal papers and conference proceedings during 2016–2021 and we summarized the usage of popular deep learning methods in Fig. 4 . The chart shows that CNN and RNN (including LSTM and GRU) are the two most dominant methods implemented in DNN-based PSSP models.

Deep learning methods and the number of PSSP works employing them during 2016–2021.
Extracting local contexts using convolutional networks. A protein secondary structure element is a local conformation formed in the polypeptide chain of the protein, composed of several amino acids. In physical space, the amino acids that make up a single secondary structure element interact with each other. From the computational point of view, these interactions, in the form of hydrogen bonds, are often invisible in the input features fed to PSSP models. Therefore, identification of local contexts/interactions on amino acid sequences needs to be done by PSSP models, and CNN has been extensively used in previous PSSP models for this purpose.
Both one-dimensional (CNN-1D) and two-dimensional CNN (CNN-2D) have been employed in previous PSSP models. CNN-1D is used more frequently since the input of a PSSP model is in the form of sequences. However, several models used CNN-2D in their architectures. CNN-2D is used in PSSP model architecture to extract temporal and spatial features of the input sequences better. Feature vectors (PSSM and one-hot encoding) of a fixed length residue window were employed in [102] , [103] , [104] , [82] as input to the two-dimensional CNN.
DeepACLSTM [105] used asymmetric convolutional filters, including two different filters, namely 1 × 2 d and k × 1 , to extract the local dependency feature of the amino acid sequence. These asymmetric convolutional filters are implemented using CNN-1D and CNN-2D. Shapovalov et al. [13] proposed a PSSP model that employed four stacked convolutional layers in its architecture. Jalal et al. [106] used multi-input CNN layers and merged the convolution outputs of each input channel.
Several studies proposed modified CNN architectures. Lin et al. [107] implemented multilayer shift and stitch in deep convolutional networks for their PSSP model to accelerate training and testing time. CNNH_PSS [108] used a highway to connect two adjacent convolutional layers to send information from the first layer to the second layer. Long and Tian [73] proposed a PSSP model with context convolutional networks, in which standard convolutional and dilated convolutional operations were joined. OCLSTM [68] optimized the parameters used in CNN using Bayesian optimization. IGPRED [66] combined CNN and graph convolutional networks.
Various architectures derived from standard CNN, such as ResNet, inception networks, U-shape networks, and fractal networks, were also employed in previous PSSP models. SPOT-1D [36] and SPOT-1D-Single [100] exploited residual networks in their architectures. Residual networks were also used in [109] . Inception networks have been utilized in several PSSP models, such as MUFOLD-SS [109] and SAINT [110] . DNSS2 [67] developed several model architectures and used various types of convolutional networks, including standard CNN, ResNet (residual block), InceptionNet (inception block), convolutional residual memory (CRM), and FractalNet. The U-shape networks have been utilized in ProteinUNet [99] .
The kernel size used in convolutional layers in a PSSP model varies, i.e., 3, 5, 7, and 11. Previous researchers determined the most appropriate kernel size by experimentation. It means that the kernel size used in PSSP models is not standardized, and different architectures may use different kernel sizes. SecNet [13] was built with stacked CNN layers. Experimentation on SecNet found that a kernel size of 7 achieved the highest prediction accuracy, and lower accuracy was achieved using larger and smaller kernel sizes. The kernel size below 7 has worse performance than that above 7. It was probably because the hydrogen bonds in the helix are formed between 3, 4, and 5 residues apart. Moreover, the average length of sheets is about six residues [111] . Although Shapovalov et al. [13] did not recommend kernel size below 7, several previous works, including [38] , [35] , used kernel sizes of 3 and 5 and achieved their best performances. Li and Yu [112] employed multiscale CNN layers with a kernel size of 3, 7, and 11. The three feature maps obtained are concatenated together as the local contextual feature vector. Although the kernel sizes mentioned above are widely used in previous PSSP models, larger kernel sizes are used in NetSurfP-2.0 [37] (129 and 257) and OPUS-TASS [56] (11, 21, 31, 41 and 51). Besides the kernel size, the number of filters used in convolutional layers in previous PSSP models also varies.
Capturing long-range interactions using RNN. In addition to identifying the local context/interactions in amino acid sequences, a PSSP model must also identify long-range interactions between amino acid residues. In sheet secondary structure, the interactions of amino acid residues (forming the hydrogen bonds) occur between distant amino acids. RNN is widely used in PSSP models to identify these long-range interactions. Furthermore, bidirectional RNNs have been used to capture forward and backward interactions. Two kinds of RNN implementation are used for this case: LSTM and GRU.
The backbone architecture of SPIDER3 [34] and SPIDER-Single [74] employed two stacked LSTM layers and two fully connected layers. Hu et al. [72] utilized ensemble learning of five base learners, and each of the base learners consisted of stacked bidirectional LSTM layers and fully connected layers. Hattori et al. [113] and Wang et al. [114] also utilized stacked LSTM layers in their PSSP model architectures. Yang et al. [115] combined bidirectional GRU and batch normalization in their PSSP model. Lyu et al. [116] built their PSSP model using two stacked bidirectional GRU layers flanked by two multi-layer-perceptron (MLP) layers. De Oliveira et al. [117] used five bidirectional GRU layers in their global classifier and five random forests in their local classifier. Ensemble learning is utilized to combine local and global classifiers.
Many PSSP models integrated both CNN and RNN in their architectures. The combination of CNN and RNN in the architecture of a PSSP model enables the PSSP model to capture both local contexts and long-range interactions.
Feature extraction using autoencoder. Besides using CNN and RNN for feature extraction, an autoencoder has been utilized to extract features on protein sequences. Several PSSP models have integrated autoencoder in their architectures [118] , [119] , [120] , [121] , [81] .
Prediction algorithms. A typical PSSP model consists of feature extraction modules and prediction modules. For the latter part, many researchers tend to use a fully connected layer and a softmax activation function. However, several PSSP studies have attempted to use different algorithms to make secondary structure predictions. Dionysiou et al. [122] , Sutanto et al. [123] , and Görmez and Aydın [124] used SVM instead of a fully connected layer. Random forest was utilized in several PSSP studies [125] , [126] , [127] , while a Bayesian classifier was used in other studies [119] , [103] , [121] .
Multitask learning. Several previous PSSP researchers designed their models to be able to perform more than one prediction task. For example, a particular model predicts not only the protein secondary structure but also other structural features, such as solvent accessibility, dihedral angles, protein disorder, and protein structural classes. Solvent accessibility measures the exposure of amino acid residues to solvent, and it is essential for understanding and predicting protein structure, function, and interactions [128] . Torsion angles ( ϕ and ψ ) provide a continuous representation of the local conformations [129] rather than the discrete secondary structures. Moreover, these continuous representations are potential for predicted local structure in fragment-free tertiary-structure prediction. Hence, it is advantageous to use protein structural properties in addition to secondary structures for protein tertiary structure prediction. Motivated by that, previous PSSP researchers performed multitask learning to predict not only the secondary structure but also other protein structural features.
Li and Yu [112] developed a PSSP model that predicts both secondary structure and solvent accessibility. SPIDER3 [34] produced several prediction outputs: secondary structure, solvent accessibility, contact number, half-sphere exposure, and dihedral angles. NetSurfP-2.0 [37] was developed using a single model to predict three-state PSSP, eight-state PSSP, relative solvent accessibility, dihedral angles, and protein disorder. MASSP [82] was designed to be able to predict both residue-level structural attributes (secondary structure, location, orientation, and topology) and protein-level structural classes (bitopic, α -helical, β -barrel, and soluble).
Ensemble learning. Ensemble learning is an approach in machine learning that combines several base classifiers/predictors to achieve better prediction performance. Previous PSSP researchers have used ensemble learning to boost the accuracy gained by their models. Hasic et al. [130] developed an ensemble of multiple artificial neural networks, where each network can have distinct parameters and architecture. eCRRNN [38] employed ten independently trained CRRNN models. PORTER5 [131] employed an ensemble of seven BRNN networks. SPOT-1D [36] leveraged the ensemble of several deep learning networks, including LSTM-BRNN and ResNet. Guo et al. [132] leveraged the ensemble learning of conditionally parameterized convolutional networks (CondGCNN) and bidirectional LSTM. Cheng et al. [126] used the ensemble learning of CNN-softmax and LSTM-random forest. AlGhamdi et al. [62] used bagging and AdaBoost with several feed-forward neural networks as the base classifiers.
Iterative learning. SPIDER3 [34] initiated iterative learning to predict protein secondary structure. The model training took four iterations, for which the earlier iteration’s output and the original input features were used as the input for the next iteration. The original input features of SPIDER3 include physicochemical properties of amino acids, PSSM, and HMM profiles.
Attention mechanism. PSSP is similar to machine translation in NLP whereby both cases use sequences as input and output. Previous PSSP models had adopted approaches and methods used in the machine translation field. One of the methods adopted by PSSP models is the attention mechanism. The first PSSP model utilizing the attention mechanism was proposed by Drori et al. [133] , where the attention mechanism proposed in [134] was used. SAINT [110] proposed a new PSSP model architecture by augmenting self-attention (transformer) [135] in Deep3I (deep inception inside inception) architecture. OPUS-TASS [56] utilized modified self-attention (transformer) by dismissing the decoder part. TMPSS [69] utilized a shared attention mechanism initially proposed in [136] for multi-way multi-language machine translation. The attention mechanism was also used in the PSSP model proposed by Guo et al. [132] .
Miscellaneous. Wang et al. [137] integrated a deep convolutional neural network with conditional random field (CRF) to model complex sequence-structure relationships and interdependency between neighboring amino acids. Instead of using real-valued neural networks, complex-valued neural networks were utilized in [140] and applied to a small training set (compact model) of the CB513 dataset. Zhao et al. [79] applied a generative adversarial networks (GAN) to perform feature extraction on the original PSSM data and fed the result into CNN. Yavuz et al. [141] proposed using a clonal selection algorithm in the PSSP model architecture to improve the data before the classification process.
Table 4 summarizes the various deep learning methods involved in the architecture of previous notable PSSP models.
Deep learning methods involved in previous notable PSSP models.
GC: graph convolutional networks; CN: convolutional neural networks; RN: residual neural networks; IN: inception networks; US: U-shape networks; FN: fractal networks; AE: auto-encoder; LS: long short-term memory; GR: gated recurrent unit; FF: feed-forward neural networks; FC: fully connected layers; CR: conditional random field; Slf: self attention (transformer); Glb: global attention.
4.3. Performance
The performance of previous notable PSSP models using hybrid features is shown in Table 5 . The performance of single-sequence-based PSSP models is shown in Table 6 .
Performance of the previous PSSP models using hybrid features including one hot encoding of amino acids, sequence profiles (PSSM, HMM profiles), and other features.
*SOV’99
Performance of the previous PSSP models using only one hot encoding of amino acids (single-sequence prediction).
Both tables show that previous PSSP models are trained on non-uniform datasets. As the training data determine the capability of PSSP models to extract patterns from the input sequences and influence the performance of the classification task, a fair performance comparison cannot be made in this setting. Moreover, using sequence profiles as input of the PSSP models may lead to an evaluation bias primarily when redundancy of profiles between the training dataset and test dataset occurs [55] . Hence, a proper comparison among PSSP models could be made if two conditions are met: (1) PSSP models are trained with identical training datasets, (2) profile redundancy between training and test dataset is diminished.
5. New approaches for feature generation
The favorable outcome of language models (LMs) in NLP has driven researchers to implement LMs in protein structure prediction. Researchers have developed protein LMs to learn the biological properties of large protein sequences in the databases [142] , [49] , [50] . The embedding features derived from these LMs carry contextual features of the amino acid residues [143] . These embedding features may replace sequence profiles for the prediction input.
Heinzinger et al. [142] adopted the bidirectional language model ELMo (Embeddings from Language Models) [144] to build a protein representation in the form of continuous vectors (embeddings) called as SecVec (sequence-to-vector). They trained ELMo on UniRef50 and assessed the predictive capability of the embeddings by applications to per-residue (word-level) and per-protein (sentence-level) tasks. For the per-residue task, they evaluated PSSP performance given one-hot amino acid encoding, SecVec embeddings, evolutionary profiles, or their combinations as inputs. SeqVec embeddings produced the best performance for prediction without evolutionary information, although it did not improve over the best existing method using evolutionary information. Regarding the computation time, generating SecVec embedding is about 60-fold faster than generating HMM profiles using HHblits.
ProtTrans [49] trained six LMs in NLP, including T5 [145] , Electra [146] , BERT [147] , ALBERT [148] , Transformer-XL [149] , and XLNet [150] on protein sequences. Five models, including ProtTXL, ProtBert, ProtXLNet, ProtAlbert, and ProtElectra, are trained using UniRef100 [151] . Besides UniRef100, ProtBert and ProtXL are also trained using BFD [152] . ProtT5 is trained on UniRef50 [151] and also trained on BFD. ProtTrans initially tokenizes the protein sequences. For the per-residue task, the token will be each amino acid. It then adds positional encoding for each respective residue. Afterward, the resulting vectors are passed through one of the ProtTrans models to create embeddings for each input token. The last hidden state of the Transformer’s attention stack of the models is used as input for the task-specific predictors (i.e., three-state PSSP model). ProtTrans experiments showed that the Q3 accuracy on CASP12 achieved by using ProtTrans embeddings is higher than using word2vec embeddings. Using the same test dataset (CASP12), the ProtTXL version is less accurate compared to an existing ELMo/LSTM-based solution (SeqVec) [142] , while all other Transformer-models outperformed the SeqVec model. Moreover, ProtTrans resulted in lower Q3 accuracy than NetSurfP-2.0 [37] , which used evolutionary information.
ESM-1b [50] used up to 250 million sequences of the UniParc database [153] , which has 86 billion amino acids. ESM-1b was trained using Transformer [135] . Transformer receives sequences of amino acids (tokens) and produces sequences of log probabilities. The output sequences of Transformer are then optimized using the masked language modeling objective. In this case, each sequence is modified by replacing a fraction of the amino acid sequence with a special mask token. The network is then trained to predict the missing tokens from the modified sequence. The purpose of using masked language modeling is to reveal the relationships/dependencies between the masked and non-masked regions. ESM-1b’s effectiveness in increasing PSSP performance was tested on the baseline PSSP model, NetSurfP-2.0. The result showed that including the ESM-1b embedding feature as NetSurfP-2.0 input substantially increases the performance of NetSurfP-2.0.
The ProtTrans and ESM-1b embedding features have been utilized to train the following PSSP models, NetSurfP-3.0 [154] , DML_SS [143] , and SPOT-1D-LM [155] . Moreover, these embedding features have been employed to train the contact map prediction model, SPOT-Contact-LM [156] , and the inter-residue distance predictor [157] .
6. AlphaFold and its impact
AlphaFold [3] became a revolution in the field of structural bioinformatics after it was able to predict 3D coordinates of protein structures with high accuracy. AlphaFold succeeded in predicting protein structures in the critical assessment of protein structure prediction round 14 (CASP14) [87] , a biennial protein structure competition held by the international community. AlphaFold can predict not only the 3D structure of single-chain proteins but also protein complexes [158] .
AlphaFold generates MSA from the genetic database search, residue pairs, and structural templates from the structural database search. Residue pairs and structural templates make up the pair representation. AlphaFold’s architecture consists of two main networks: Evoformer and the structure module. Evoformer consists of 48 blocks containing attention-based and non-attention-based components. Each block of the Evoformer updates the MSA and the pair representation and facilitates communication between them. Evoformer performs three steps to update the MSA representation: row-wise gated self-attention, column-wise gated self-attention, and transition. The first step incorporates information from the pair representation as input to synchronize the MSA with the updated pair representation from the previous block. For each step, the output is added to the input and the result of this addition is fed into the next step. The result of the MSA update is then injected into the pair representation.
Evoformer utilizes a triangle portrayal to depict the pair representation into a 3D structure. This triangle is a graph of three different nodes. The nodes represent amino acid residues, whereas the edges represent the connectivity between amino acid residues. Evoformer performs several operations to update the pair representation, including: injection of the updated MSA into the pair representation by executing the outer product mean operation, two non-attentional triangle multiplicative updates (using ‘outgoing’ edges and ‘incoming’ edges), two attentional triangle updates (self-attention around starting node and self-attention around terminating node), and transition.
Each block of the Evoformer is followed by a structure module. The objective of the structure module is to generate the 3D representation of the input protein by using the MSA and the pair representation that the Evoformer has manipulated. Moreover, the structure module also takes backbone frames as input. The backbone frames represent each residue as one free-floating unit of the protein polypeptide backbone. The geometry of N-C α -C of each amino acid residue plays an important part that makes up the actual polypeptide backbone, whereas the polypeptide backbone exhibits the 3D structure of the protein. The structure module consists of three parts: Invariant Point Attention (IPA), relative rotation and translation predictors, and side-chain angles predictor. The IPA is a geometry-aware attention operation that updates the MSA’s single representation, which is then used to update the backbone frames via rotation and translation operations. Together with the updated backbone frames, the single representation of the MSA predicts the side-chain angles and the position of atoms composing each residue.
The success of AlphaFold in predicting the 3D structure of proteins is induced by the combination of bioinformatics and physical approaches it implemented. Evoformer successfully produces representations that reveal spatial and evolutionary relationships of the residues. Moreover, these spatial and evolutionary relationships are used by the structure module to capture the orientation of the protein backbone, thus the 3D structure of the protein can be revealed. With the invention of AlphaFold, the prediction of the 3D structure of proteins from their amino acid sequences is already practicable. It directly predicts the tertiary structure of proteins by using only their primary structures, bypassing the intermediary tasks in protein structure prediction, such as PSSP, folds prediction, and prediction of protein structural features.
This advancement in protein structure prediction pioneered by AlphaFold undoubtedly raises the question of whether PSSP and the prediction of protein structural features are still necessary since predicting protein tertiary structure from its amino acid sequence becomes practical. AlphaFold is powerful in predicting protein tertiary structures; however, since AlphaFold relies on MSA, it is particularly beneficial for homologous proteins. AlphaFold performance is imperfect in predicting the structure of proteins with no prior evolutionary information (proteins without known homologs) [159] , [160] , whereas proteins without known homologs are currently estimated at 20% of all metagenomic protein sequences [161] and about 11% of eukaryotic and viral proteins [162] . This case of low-quality MSA will lead to less accuracy in structure prediction. Moreover, leveraging MSA requires high computing resources, especially to generate MSA from massive collections of protein sequences from multiple protein databases. PSSP with evolutionary information as input may fall for the same problem. Here, PSSP and prediction of protein structural features, especially ones that do not rely on evolutionary information might still be needed for proteins without known homologs.
7. PSSP in post-AlphaFold publication
Although AlphaFold is a breakthrough in protein structure prediction due to its highly accurate prediction, we found that PSSP studies continue after the release of AlphaFold. This section discusses the PSSP models proposed by researchers after AlphaFold was published (2021). PSSP studies in the post-AlphaFold era focus on one or more objectives including: (1) taking MSA instead of PSSM as direct input to the PSSP model, (2) improving the quality of sequence profiles, (3) exploiting protein language models (LMs) as alternative input features, and (4) other objectives.
7.1. Taking MSA as direct input to the PSSP model
AlphaFold has successfully used MSA instead of sequence profiles to produce accurate structure prediction. Another study by Ju et al. [163] suggested the use of MSA as a direct input for the PSSP model concerning that sequence profiles derived from MSA (PSSM and HMM profiles) may not be able to represent residue mutations and correlations. The model, called Seq-SetNet, performed two stages, encoding and aggregation, to deduce the structural properties of each amino acid in the input sequence. The encoding module processes each homologue sequence from the MSA independently and produces a contextual vector in which each position in the vector covers the information of its surroundings. The encoding module is constructed of 1D convolutional residual networks with 8 residual blocks. The aggregation module aggregates the outputs of the encoder module, namely the contextual features extracted from all the homologous proteins. The element-wise maximum function is applied to the contextual features of the homologue proteins to perform the aggregation. The experiment result showed that Seq-SetNet outperformed the baseline PSSP model, which used handcrafted features, such as PSSM and HMM profiles.
7.2. Improving the quality of sequence profiles
Considering the low-quality sequence profiles that proteins without known homologs may have, a recent PSSP study by Wang et al. [164] proposed PSSM-Distil, which conducts knowledge distillation to produce enhanced PSSM leveraging student–teacher networks. PSSM-Distil basically performs several steps. First, it trains the teacher network with high-quality PSSM. Secondly, it down-samples the high-quality PSSM to get the low-quality one by using prior statistics and then, the low-quality PSSM and pre-trained BERT are used to train a network producing an enhanced PSSM. Thirdly, it performs secondary structure classification using enhanced PSSM on a student network. It also performs the secondary structure classification on the teacher network. A loss function based on the loss of the student network and the loss of the teacher network is calculated to measure the PSSP performance. The experiment result showed that using PSSM-Distil instead of the standard PSSM increased the accuracy of PSSP in low-quality PSSM cases in BC40 and CB513 datasets. Subsequently, the same researchers also proposed a dynamic scoring matrix (DSM)-Distil to replace PSSM and other widely used features [165] . This feature leveraged the pre-trained BERT to construct a dynamic scoring matrix (DSM) and performed knowledge distillation on the DSM.
7.3. Exploiting protein language models (LMs) as input features
Following the success of language models in NLP, protein language models (LMs) have appeared to be an alternative to replace the evolutionary information as an input feature for PSSP models. DML_SS [143] utilized embedding features derived from the ProtTrans language model (ProtT5-XL-U50). DML_SS used a deep centroid model based on deep metric learning in its model architecture. This deep centroid model is used to learn deep embedding to keep the samples with the same labels close to each other in the embedding space while samples with different labels remain distant from each other. DML_SS employed embedding networks consisting of inception networks, convolutional networks, and fully connected layers. DML_SS proved that by using embedding features, its prediction accuracy in the CB513 test dataset is higher than if it used hybrid features (sequence profile features and physicochemical properties).
LMs have also been utilized to improve the performance of NetSurfP-2.0. NetSurfP-3.0 [154] employed the architecture of NetSurfP-2.0 [37] and utilized latent sequence representation derived from the ESM-1b as the input feature. NetsurfP-3.0 showed that it surpasses the performance of NetSurfP-2.0 in CB513 and TS115 test datasets, although it does not perform better on the CASP12 dataset. Moreover, using ESM-1b embedding, NetSurfP-3.0 runs around 600 times faster than NetSurfP-2.0.
SPOT-1D-LM [155] combined amino acid one-hot encoding and embedding features from ProtTrans and ESM-1b. SPOT-1D-LM trained an ensemble of three different model architectures, including a two-layer LSTM, a multi-scale ResNet, and a multi-scale ResNet-LSTM. SPOT-1D-LM has successfully raised the Q3 accuracy of single-sequence-based PSSP in the TEST2018 dataset to higher than 80%, namely 86.74% using the ensemble model. It is a great leap in the Q3 accuracy compared to single-sequence-based prediction using the same test dataset. SPOT-1D-LM also outperformed other single-sequence-based PSSP models in terms of Q8 accuracy in the same test dataset.
Table 7 shows the performance of LM-based PSSP models. Similar to Table 5 and Table 6 cases, a fair comparison of LM-based PSSP models can be conducted when the models are trained using identical training datasets.
The performance of PSSP models using language models (LMs).
7.4. PSSP studies aiming at other objectives
The length of input sequences fed into DNN-based PSSP models may vary due to the different amino acid compositions of the input proteins. Thus, padding is a common practice conducted to have equal size inputs passed into the networks. Padding is done by adding zero values into the sequences shorter than a pre-determined size such that they are adjusted to that size. However, this standard practice raises an issue: the networks will change the feature vectors corresponding to the padded positions to non-zero vectors during forward propagation, affecting the prediction result of the non-padded positions. To address this issue, Yang et al. [166] proposed a modified 1D batch normalization by exploiting a mask matrix, so that padding positions in the feature vectors do not participate in normalization operations. Hence, the feature vectors in these padding positions remain all-zeros. Furthermore, they [166] also proposed a PSSP model with architecture comprised of CNNs and fully connected layers along with the implementation of masked batch normalization.
Stapor et al. [167] proposed lightweight ProteinUnet2, a PSSP model based on U-net convolutional networks. ProteinUnet2 takes sequence profiles (PSSM and HMM profiles) and SPOT-Contact features as inputs. ProteinUnet2 is developed based on the architecture of ProteinUnet [99] , extended with multiple inputs using multiple contractive paths. The experiment shows that ProteinUnet2 achieves a comparable prediction accuracy to SPOT-1D and SAINT. However, ProteinUnet requires less training and prediction time than SPOT-1D and SAINT. Hence, it could be useful for a system with low computing resources. Moreover, ProteinUnet2 can accelerate the secondary structure prediction of large datasets. They [167] also used the Adjusted Geometric Mean (AGM) [168] metric to assess the performance of PSSP models, especially due to the imbalanced dataset available in the field.
Görmez and Aydın [169] proposed IGPRED_Multitask to predict several tasks, including protein secondary structure, solvent accessibility, and torsion angles. The architecture of IGPRED_Multitask utilizes graph neural networks, CNN, and bidirectional LSTM. They [169] also attempted hyper-parameter optimization using Bayesian optimization. Besides one hot encoding of amino acids and their physicochemical properties, IGPRED_Multitask also took five scores of structural profiles as additional input features. IGPRED_Multitask was trained using the same training set as OPUS-TASS [56] . It showed its outstanding performance compared to OPUS-TASS on several benchmark test datasets, including TEST2016, TEST2018, CAMEO93, CAMEO93_HARD, CASP12, CASP13, CASPFM, and HARD68.
8. Summary and outlook
PSSP has been a mature research topic in structural bioinformatics. The three-state PSSP has achieved accuracy close to its theoretical limit, 88–89% [170] . However, considering the current protein structure databases, a new study reported that the theoretical limit of PSSP can be extended to 94% [171] . This means that there is still an accuracy gap to fill.
The state of the art of PSSP accuracy is not distributed evenly for all classes (helix/sheet/coil). The prediction accuracy of sheet and coil is not as high as that of helix. The ability to precisely capture long-range interactions, especially in sheet ( β -strand), may need to be improved to boost the prediction accuracy, even though previous models had used RNN to handle this problem. Recently, the embedding features derived from protein language models can reveal the contextual information of the amino acid residues [143] . Using embedding features derived from protein language models in NetSurf-P3.0, DML SS, and SPOT-1D-LM has successfully improved the prediction accuracy of the benchmark test datasets. The impact of embedding features on the ability to accurately predict non-helix structures can be further investigated.
On the other side, there is room for improvement in both eight-state PSSP and single-sequence-based PSSP, as prediction accuracy is still limited. The challenge in the eight-state PSSP was the imbalanced samples of each class; some classes lacked samples, such as B, S, G, and I. The eight-state PSSP model performance can be affected by this imbalanced dataset issue. Thus, the methods and techniques to train DNN on this imbalanced dataset case are potential for future work. A new classification of four and five-state PSSP proposed by Shapovalov et al. [13] may become a potential alternative to PSSP.
DNN has been employed in single-sequence-based PSSP, but only a few models have been published. Various deep neural network architectures are feasible to be implemented. Adopting previous DNN-based PSSP models into single-sequence cases is also worth trying. Considering the recent success of LM-based PSSP models in enhancing the PSSP accuracy, combining LM-derived features and amino acid sequences are potential input features for future PSSP models. Furthermore, the issue remaining in the post-AlphaFold era is the low-quality evolutionary information of the proteins without known homologs. Hence, the structure prediction models that exclude evolutionary information will be useful for such proteins. Single-sequence-based PSSP models can be further developed.
CRediT authorship contribution statement
Dewi Pramudi Ismi: Conceptualization, Investigation, Methodology, Writing – original draft, Writing – review & editing. Reza Pulungan: Conceptualization, Methodology, Writing – review & editing. Afiahayati: Conceptualization, Analysis, Methodology, Writing – review.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work is supported in part by the Directorate of Research, Technology, and Community Services, Directorate General of Higher Education, Research, and Technology, Ministry of Education, Culture, Research, and Technology of the Republic of Indonesia, through Doctoral Dissertation Research Grant No. 1906/UN1/DITLIT/Dit-Lit/PT.01.03/2022.
- Help & FAQ
A Short Review on Protein Secondary Structure Prediction Methods
- Biochemistry & Molecular Biology
Research output : Chapter in Book/Report/Conference proceeding › Chapter (Book) › Other › peer-review
This chapter discusses seven protein secondary structure prediction methods, covering simple statistical-and pattern recognition-based techniques. The prediction methods include Chou-Fasman, Garnier, Osguthorpe and Robson (GOR), PHD, neural network (NN)-based protein secondary structure prediction (PSIPRED), SPINE-X, protein secondary structure prediction (PSSpred) and meta methods. The chapter assesses the performance of different methods using the Q3 measure. It investigates the accuracy of secondary structure prediction for target proteins by the alignment/threading programs. The top-performing methods, for example, PSSpred, PSIPRED, and SPINE-X, are consistently developed using NNs, which suggests that NNs are one of the most suitable pattern recognition algorithms to infer protein secondary structure from sequence profiles. It is found that the secondary structure prediction by the alignment/threading methods that combined PSIPRED with other informative structural features, such as solvent accessibility and dihedral torsion angles, was more accurate.
- Chou-Fasman method
- Garnier, Osguthorpe and Robson method
- Pattern recognition algorithms
- Protein secondary structure prediction
Access to Document
- 10.1002/9781119078845.ch6
Other files and links
- Link to publication in Scopus
T1 - A Short Review on Protein Secondary Structure Prediction Methods
AU - Yan, Renxiang
AU - Song, Jiangning
AU - Cai, Weiwen
AU - Zhang, Ziding
PY - 2015/12/28
Y1 - 2015/12/28
N2 - This chapter discusses seven protein secondary structure prediction methods, covering simple statistical-and pattern recognition-based techniques. The prediction methods include Chou-Fasman, Garnier, Osguthorpe and Robson (GOR), PHD, neural network (NN)-based protein secondary structure prediction (PSIPRED), SPINE-X, protein secondary structure prediction (PSSpred) and meta methods. The chapter assesses the performance of different methods using the Q3 measure. It investigates the accuracy of secondary structure prediction for target proteins by the alignment/threading programs. The top-performing methods, for example, PSSpred, PSIPRED, and SPINE-X, are consistently developed using NNs, which suggests that NNs are one of the most suitable pattern recognition algorithms to infer protein secondary structure from sequence profiles. It is found that the secondary structure prediction by the alignment/threading methods that combined PSIPRED with other informative structural features, such as solvent accessibility and dihedral torsion angles, was more accurate.
AB - This chapter discusses seven protein secondary structure prediction methods, covering simple statistical-and pattern recognition-based techniques. The prediction methods include Chou-Fasman, Garnier, Osguthorpe and Robson (GOR), PHD, neural network (NN)-based protein secondary structure prediction (PSIPRED), SPINE-X, protein secondary structure prediction (PSSpred) and meta methods. The chapter assesses the performance of different methods using the Q3 measure. It investigates the accuracy of secondary structure prediction for target proteins by the alignment/threading programs. The top-performing methods, for example, PSSpred, PSIPRED, and SPINE-X, are consistently developed using NNs, which suggests that NNs are one of the most suitable pattern recognition algorithms to infer protein secondary structure from sequence profiles. It is found that the secondary structure prediction by the alignment/threading methods that combined PSIPRED with other informative structural features, such as solvent accessibility and dihedral torsion angles, was more accurate.
KW - Chou-Fasman method
KW - Garnier, Osguthorpe and Robson method
KW - Pattern recognition algorithms
KW - PHD method
KW - Protein secondary structure prediction
KW - SPINE-X
UR - http://www.scopus.com/inward/record.url?scp=84978216552&partnerID=8YFLogxK
U2 - 10.1002/9781119078845.ch6
DO - 10.1002/9781119078845.ch6
M3 - Chapter (Book)
AN - SCOPUS:84978216552
SN - 9781118893685
BT - Pattern Recognition in Computational Molecular Biology
A2 - Elloumi, Mourad
A2 - Iliopoulos, Costas S.
A2 - Wang, Jason T. L.
A2 - Zomaya, Albert Y.
PB - Wiley-Blackwell
- Open access
- Published: 14 June 2006
Improving the accuracy of protein secondary structure prediction using structural alignment
- Scott Montgomerie 1 ,
- Shan Sundararaj 1 ,
- Warren J Gallin 1 &
- David S Wishart 1 , 2
BMC Bioinformatics volume 7 , Article number: 301 ( 2006 ) Cite this article
98 Citations
Metrics details
The accuracy of protein secondary structure prediction has steadily improved over the past 30 years. Now many secondary structure prediction methods routinely achieve an accuracy (Q3) of about 75%. We believe this accuracy could be further improved by including structure (as opposed to sequence) database comparisons as part of the prediction process. Indeed, given the large size of the Protein Data Bank (>35,000 sequences), the probability of a newly identified sequence having a structural homologue is actually quite high.
We have developed a method that performs structure-based sequence alignments as part of the secondary structure prediction process. By mapping the structure of a known homologue (sequence ID >25%) onto the query protein's sequence, it is possible to predict at least a portion of that query protein's secondary structure. By integrating this structural alignment approach with conventional (sequence-based) secondary structure methods and then combining it with a "jury-of-experts" system to generate a consensus result, it is possible to attain very high prediction accuracy. Using a sequence-unique test set of 1644 proteins from EVA, this new method achieves an average Q3 score of 81.3%. Extensive testing indicates this is approximately 4–5% better than any other method currently available. Assessments using non sequence-unique test sets (typical of those used in proteome annotation or structural genomics) indicate that this new method can achieve a Q3 score approaching 88%.
By using both sequence and structure databases and by exploiting the latest techniques in machine learning it is possible to routinely predict protein secondary structure with an accuracy well above 80%. A program and web server, called PROTEUS, that performs these secondary structure predictions is accessible at http://wishart.biology.ualberta.ca/proteus . For high throughput or batch sequence analyses, the PROTEUS programs, databases (and server) can be downloaded and run locally.
The field of protein structure prediction began even before the first protein structures were actually solved [ 1 ]. Secondary structure prediction began [ 2 , 3 ] shortly after just a few protein coordinates were deposited into the Protein Data Bank [ 4 ]. In the 1980's, as the very first membrane proteins were being solved, membrane helix (and later membrane β-strand) and signal peptide prediction methods began to proliferate [ 5 ]. Homology modeling, as a way of predicting 3D structures, followed in the mid 1980's [ 6 ]. Later, in the 1990's the concept of threading (both 2D and 3D) emerged, thereby allowing reasonably accurate fold prediction to be performed on very distantly related sequences [ 7 , 8 ]. Over time, the accuracy and reliability of most protein structure prediction methods has grown considerably. This is, in part, due to the development of more sophisticated prediction methods using neural nets or Hidden Markov Models [ 9 ], the development of more rigorous testing methods [ 10 , 11 ] and the explosive growth in both sequence and structure data on which scientists can "train" their software (35,000+ structures and 2,000,000+ sequences).
Protein structure prediction continues to be an actively developing field with more than 400 papers being published on the subject each year. Incremental improvements in prediction accuracy are still being reported and until "the protein folding problem" is formally solved, it is likely that protein structure prediction will continue to be an active area of research and development [ 12 ]. The continuing improvements in structure prediction accuracy are also having an effect on how proteins are analyzed and annotated. While once an anathema to most protein chemists, secondary structure prediction is now becoming a routine part of many protein analyses and proteome annotation efforts [ 13 ]. Annotation systems such as PEDANT [ 16 ], BASYS [ 14 ], BACMAP [ 17 ], PSORTB [ 15 ] and others all depend on large scale secondary structure predictions to assist in identifying possible functions, to determine subcellular locations, to assess global trends in secondary structure content among different organisms or certain organelles, to identify protein folds or to enumerate fold classes (all alpha, all beta, mixed), to identify domains, and to estimate the proportion of "unfolded" or unstructured proteins in a given genome [ 20 – 22 , 27 – 29 ]. Likewise protein secondary structure predictions can play a valuable role for molecular biologists in deciding where and how to subclone protein fragments for expression (i.e. where to cut the gene), where to join or insert gene fragments, or in choosing where to add affinity tags for protein purification [ 26 , 27 ]. Secondary structure predictions can also be used to calibrate CD and FTIR measurements when monitoring the folding or unfolding proteins with no known 3D structure [ 19 , 18 ]. Secondary structure predictions may also be used to assist in the assignment of NMR spectra (of known or novel proteins), to re-reference chemical shifts and to help determine protein flexibility [ 23 , 24 ].
Currently the performance (or Q3 score) of the best secondary structure prediction methods, such as PSIPRED [ 35 ], JNET [ 36 ] and PHD [ 13 , 37 ] is between 75–77%. These methods, which are specific to water-soluble proteins, utilize BLAST or PSI-BLAST searches of the non-redundant protein sequence database to obtain evolutionary information. This information is then fed through a multi-layered feed-forward neural network that has previously been trained on known structures and known alignments to learn characteristic sequence/structure patterns. Those patterns are then used to predict the secondary structure of the query protein [ 38 ]. Similarly good scores can also be achieved using Hidden Markov Models with programs such as SAM-T02 [ 39 ]. More recently approaches that combine multiple high quality methods (a "jury of experts" or meta methods) have been described [ 40 , 41 ] and these appear to do even better than the single-pass prediction approaches.
What is somewhat surprising about the methods described so far is that they do not fully exploit the information that is available in the protein structure databases. So far as we are aware, none of the above-mentioned methods attempt to find sequence homologues in the PDB and to use the known secondary structure of those homologues to assign, map or predict the secondary structure of the query protein. As a rule, this sequence/structure alignment approach to secondary structure assignment is normally reserved for homology modeling programs [ 7 , 42 ]. For pairwise sequence identities of >35%, these secondary structure mappings are typically more than 90% accurate. However, we believe this 3D-to-2D mapping approach to general secondary structure prediction is not being fully exploited. A recent survey has found that less than 3% of new protein structures deposited into the PDB have a totally novel fold [ 43 ]. Even among structural genomics projects, where novel folds are explicitly being sought and solved, less than 10% of the targets exhibit completely novel folds [ 44 , 45 ]. Furthermore, we have found that nearly 3/4 of newly deposited PDB structures have sequence identities greater than 25% to a pre-existing structure. In other words, the vast majority of newly solved proteins could have at least a portion of their secondary structures predicted via this simple 3D-to-2D mapping approach. Thus, by combining a PDB-based structure alignment with a high quality de novo structure prediction program it may be possible to achieve a much higher overall Q3 score for protein secondary structure prediction.
Here we wish to describe a program, called PROTEUS, that exploits this concept of 3D-to-2D mapping and integrates it with multiple de novo methods to accurately predict protein secondary structure. Specifically, PROTEUS achieves an average Q3 score of 88% when tested on newly solved protein structures. This level of accuracy is 12–15% above that previously reported [ 35 – 41 ]. If a query protein has at least some portion of its sequence that is homologous to an existing PDB structure, the average Q3 score exceeds 90%. If absolutely no homology is found, or if the 3D-to-2D mapping option is turned off, the average accuracy of this method is still above 79%. In addition to greatly improving the average performance of secondary structure prediction, we have parallelized the prediction algorithm, developed a simple installation protocol and made the full source code and all associated databases freely available and as portable as possible. This was done in an effort to facilitate proteome annotation and to encourage large scale pipelined analyses or proteome-wide structure predictions to be done locally rather than remotely.
Implementation
PROTEUS consists of three components: 1) a large (12,464 entries), non-redundant and continuously updated database of sequences with known secondary structures; 2) a multiple sequence alignment algorithm for secondary structure mapping and homology prediction and 3) a "jury-of-experts" secondary structure prediction tool consisting of three different, high-performing de novo secondary structure prediction programs (PSIPRED, JNET and a home-made tool called TRANSSEC). The prediction algorithm itself involves four steps including an initial search against the PDB sequence database to determine if all or part of the query sequence is similar to a known structure. If a hit is found, a secondary structure mapping is performed on whatever component that mapped to the query. In the second step, a de novo secondary structure prediction using our three different (JNET, PSIPRED and TRANSSEC), high quality neural network (NN) approaches is performed. In the third step these three NN predictions are then fed as inputs into a fourth neural network, which then combines these predictions to make a prediction of its own (i.e. a decision by a jury of experts). Finally, the jury-of-experts prediction and the results of the initial homology search are combined to produce the final secondary prediction for PROTEUS (see Figure 1 ). Combining the two prediction methods allows PROTEUS to fill in any prediction gaps derived from the initial 3D-to-2D mapping process and always yields a full sequence prediction, regardless of the extent of sequence overlap to a PDB hit.

A) Flow chart outlining how protein sequences are processed by PROTEUS. Each query sequence is simultaneously processed through PROTEUS' three de novo predictors (lower left corner) and through a BLAST comparison and global alignment (via XALIGN) against the PDB, to yield a 3D-to-2D mapping. The two secondary structure predictions are merged and filtered to produce a final consensus prediction. B) Detail illustrating how the two predictions are merged in the final processing step.
Key to the success of PROTEUS is its effective use of secondary structure databases. PROTEUS' secondary structure database (PROTEUS-2D) is assembled from a non-redundant version of the Protein Data Bank (PDB) in which all sequences with >95% sequence identity to any other sequence were removed using the CD-HIT utility [ 47 ]. Each sequence was then assigned a secondary structure using VADAR [ 48 ]. The secondary structures were then checked and filtered so as not to contain "impossible" structures, such as sheets or helices containing a single residue. VADAR uses a consensus method of identifying secondary structures that closely matches "simplified" DSSP [ 49 ] structure assignments (8 state to 3 state), STRIDE [ 50 ] and generally agrees well with manual secondary structure assignments made by X-ray crystallographers and NMR spectroscopists. In fact, using the PROTEUS-2D database of secondary structures, the performance of PSIPRED and JNET was actually found to improve slightly over the performance quoted for DSSP-assigned secondary structures (77% vs. 75%). The secondary structure content of the PROTEUS-2D database, which currently contains over 2.2 million residues from more than 12,400 sequences, is 33% helix, 29% beta sheet and 38% coil. Because of its critical importance to the prediction process, the entire PROTEUS-2D database is automatically updated on a weekly basis. This database is also freely available for download at the PROTEUS website.
The PDB homology search and 3D-to-2D mapping process in PROTEUS both employ BLAST (using the default BLOSUM 62 scoring matrix and standard gap penalty parameters) to score and align high scoring hits found in the PROTEUS-2D database. Those database sequences having an expect score greater than 10 -7 to the query sequence are retained for further analysis. This optimal expect value was determined by extensive testing with cut-offs ranging from 10 -1 to 10 -15 . Depending on the length and domain structure of the query sequence up to 20+ homologues may be identified by this process. The pairwise BLAST alignments are then used to assemble a multiple sequence alignment over the length of the query sequence. The resulting multi-sequence alignment is then used to directly map the secondary structure of the PROTEUS-2D database sequences (or a portion thereof) to the query sequence. The mapping process involves sliding a 7 residue window over each aligned sequence and assigning a similarity score (based on the sequence identity over that 7 residue window to the query sequence) to the central residue. The sequence with the highest "identity score" for any given residue is then privileged to assign its secondary structure to the aligned residue in the query sequence. In this way the secondary structure of the query sequence is essentially predicted by homology. For those query sequences that are predicted in this manner (with more than 95% sequence coverage), PROTEUS also produces an image of the approximate 3D fold using the PDB coordinates to generate the picture.
In situations where no homologue is found, or only a portion of the query sequence could be predicted by 3D-to-2D mapping (as might be found in multi-domain proteins), PROTEUS resorts to a jury-of-experts prediction to cover the unpredicted portion. This jury-of-experts approach uses three neural net predictors: PSIPRED [ 35 ], JNET [ 36 ] and our own TRANSSEC (Q3 = 70%, SOV = 73%) methods. The results from these predictors are then fed into a fourth neural network to produce a consensus prediction in a manner similar to that described previously [ 40 ].
The methods and underlying theory to PSIPRED and JNET have been published previously and the programs were used as received without further modification. The TRANSSEC program was developed in-house using a Java-based neural network package known as Joone [ 51 ]. TRANSSEC's underlying approach is relatively simple, consisting of a standard PSI-BLAST search integrated into a two-tiered neural network architecture. The first neural network operates only on the sequence, while the second operates on a 4 × N position-specific scoring matrix consisting of the secondary structure determined via the first network. The first neural net uses a window size of 19, and was trained on 1000 sequences from the PROTEUS-2D database (independent from those used in training the other neural nets). This neural net had a 399-160-20-4 architecture (21 × 19 inputs, 2 hidden layers of 160 and 20, and four outputs) and typically predicts the secondary structure of any given protein with a Q3 = 64–65%. TRANSSEC's neural net secondary structure predictions are performed on all PSI-BLAST homologues to the query sequence These homologues are then multiply aligned using XALIGN [ 46 ] with the secondary structure serving as a guide to place gaps and insertions. The resulting secondary structure-based alignment (and corresponding confidence scores) is then used as input for a second neural network. TRANSSEC differs from most other prediction programs (PHD, PSIPRED) in that the predicted secondary structure, instead of the sequence, is used as input for the second neural network. What TRANSSEC attempts to do is to learn, via a neural net, how to "average" aligned secondary structures in a more intelligent way. A simple averaging of secondary structures typically reduces the prediction accuracy from 65% (for a single prediction) to 63% (for the averaged prediction), while using a neural net increases the performance by about 7% over naive averaging. The second neural net in TRANSSEC was trained on 1000 sequences from the PROTEUS-2D database, and achieved a Q3 score of 70% and a SOV score of 72%. It used a window size of 9, and was based on a 36-44-4 architecture.
The jury-of-experts program, (JOE) which combined the results of the three stand-alone secondary structure predictions was also developed using Joone. JOE consisted of a standard feed-forward network containing a single hidden layer. Using a window size of 15, the structure annotations and confidence scores from each of the three methods (JNET, PSIPRED, and TRANSSEC) were used as input. The JSP neural net was trained and tested (using a leave-one-out approach) on 100 sequences chosen randomly from the non-redundant database mentioned above. Four output nodes were used, one for each of helix, strand or coil, as well as a fourth denoting the beginning and end of the sequence. A back-propagation training procedure was applied to optimize the network weights. A momentum term of 0.2 and a learning rate of 0.3 were used, and a second test set of 20 proteins was applied at the end of each epoch, to ensure that the network was trained for the most optimal number of iterations. The JOE program outputs not only the secondary state call (H for helix, C for coil and E for beta strand), but also a numeric confidence score (ranging from 0 to 9, with 9 being most confident). Relative to simple averaging, the JOE program is able to improve secondary structure predictions by an average of 3% (79.1% vs. 76.4%). The improvement achieved using this jury of experts approach is likely due to the fact that JNET, PSIPRED and TRANSSEC perform differently for different types of proteins, with one method typically outperforming the other two depending on the secondary structure content, protein length or amino acid content. It appears that JOE's neural net was able to learn which method or which segmental prediction to trust more and therefore to place more weight on those predictions. It also appears that the JOE method also learned to modify the JNET and PSIPRED predictions (typically by lengthening them) to conform better to the VADAR-assigned secondary structures.
The final step in the PROTEUS algorithm involves merging the homology prediction (if available) with the jury-of-experts predictions. The PROTEUS-merge program was designed to accommodate three situations: 1) the case where no PDB homologue could be found, 2) the case where complete 3D-to-2D mapping was achieved and 3) the case where the 3D-to-2D mapping provided only partial coverage of the full query sequence. In the simple situation where no 2D-to-3D prediction is available (Case 1), the merge process simply takes the jury-of-experts or de novo result. Similarly, if a complete PDB-based secondary structure prediction is available (Case 2), the jury-of-experts prediction is generally ignored. In particular, if the homologue confidence score is equal to or greater than the consensus de novo score, then the homologue structure assignment is retained. Otherwise the de novo structure assignment is kept. Typically the de novo confidence scores range from 3–9, while the homologue confidence scores range from 8–9. The confidence of a homologue prediction is based on the running average (over a 7 residue window) of the sequence identity between the query sequence and that of the top matching PDB homolog. If the sequence identity is less than 30% (or 2/7), the confidence score assigned to the middle residue in the window is 8. If it is greater, the confidence score of the middle residue is 9. Confidence scores for the consensus de novo predictions are determined by the weightings of specific neural network nodes. If a homologous sequence or a group of homologous sequences is found (as with multi-domain proteins) that did not cover the entire length of the query sequence (Case 3), the unpredicted or unmapped portion is assigned the secondary structure determined by our Jury-of-experts approach (Figure 1 ).
PROTEUS also has a number of I/O utilities and interface tools that allow it to accept protein sequences (in FASTA and Raw format) and to produce colorful and informative output including all sequence alignments, corresponding BLAST scores, sequence matches, confidence scores, colored secondary structure annotation as well as 3D images of any modeled structures. Additional data handling and task handling tools were also written to manage the server side of the program, to update the PROTEUS-2D database on a weekly basis, and to parcel out tasks to other processors in a parallel fashion. The programs used to create PROTEUS and the PROTEUS web server were written in both C and Java 1.5. Specifically, XALIGN, VADAR, JNET, BLAST and PSIPRED were written in ANSI standard C, while TRANSSEC, the Jury-Selector, most of the input/output handling routines, as well as the web server interface were written in Java. The PROTEUS-2D update script was written in Perl.
PROTEUS' performance was tested in four different ways, 1) through leave-one-out testing on a set of 100 training proteins from the PROTEUS-2D database; 2) through a "blind" test and comparison on the latest EVA training set (1644 proteins); 3) through analysis of 125 randomly chosen proteins that were recently solved by X-ray and NMR; and 4) through direct comparisons of 10 randomly chosen proteins to well-known secondary structure web servers. The intent of these different tests was to gain some understanding of the performance of PROTEUS under different prediction situations and to assess its performance relative to other well known predictors. For the first test, the performance of the jury-of-experts system was assessed using a leave-one-out strategy on 100 randomly chosen proteins form the PROTEUS-2D databases. As previously mentioned, this method achieved a Q3 score of 79.1% and a SOV score of 77.5%. When this method was combined with the 3D-to-2D mapping (excluding identical matches from the PROTEUS-2D database), the performance was Q3 = 88.0% and SOV = 86.5%. The performance for the "full" version of PROTEUS ( de novo plus homologue mapping) is about 10–15% higher than previously reported for other methods. Because this first test was done on training data (albeit using a leave-one-out strategy) it might be argued that the high performance may be due to overtraining or to the small sample size.
To more legitimately assess the performance of PROTEUS a second "blind" test was done on data not part of PROTEUS' training set and for which no PDB homologues would be expected. Specifically the most recent release (March 2006) of the EVA [ 11 ] sequence-unique subset of the PDB was downloaded and used to measure the performance of PROTEUS. The EVA collection represent a set of non-homologous proteins that do not match any 100+ residue segment of any other protein in the PDB with greater than 33% sequence identity. The EVA test-set has been used for a number of years to benchmark protein secondary structure predictors, particular for CASP competitions [ 11 ]. The use of a sequence-unique data set such as EVA is intended to simulate the situation where one might be predicting secondary structures in a structural genomics project, where novel fold identification is key. In this particular situation one would expect that the PROTEUS predictions would be dominated by its de novo methods and that the Q3 and SOV scores would be somewhat reduced over the first test. A total of 1644 protein sequences and PDB ID's were obtained from the EVA website and the secondary structure for each of the test-set proteins was assigned by VADAR [ 48 ]. PROTEUS was then used to predict the secondary structures and the performance was evaluated against the VADAR-assigned secondary structures. The program was tested in two modes, one with the PDB homologue search turned off ( de novo prediction only) and other with the PDB search turned on. In both cases the Q3 and SOV scores were calculated for each protein in the 1600 protein test set. Note that the SOV score is similar to Q3 but more sensitive to the segment grouping or overlap of secondary structure elements [ 52 ]. At the same time the Q3 and SOV scores for JNET (alone) and PSIPRED (alone) were also determined for all 1644 EVA proteins. Additionally the secondary structure predictions posted on the EVA server for PORTER [ 31 ], PROF-KING [ 32 ], PROFSEC [ 34 ], SAM-T99-sec [ 33 ] and VASPIN [ 30 ] were also downloaded and processed in a similar manner to the PSIPRED and JNET predictions. Note that the number of predictions for these predictors was much less than 1644 as the EVA server often only performs a small number (<200) of predictions for any given predictor. As seen in Figure 2 , PROTEUS achieves a Q3 of 77.6% (SOV = 78.2%) when its homologue search is turned off and a Q3 score of 81.3% (SOV = 81.8%) when the homologue search is turned on (with the exact match in the PDB removed), with a standard deviation of 11.0% and 14.1%, respectively. Evidently, even in a sequence-unique data set, some fragmentary homology is still detectable by PROTEUS. In particular for those proteins that exhibited some detectable homology to a portion of a PDB structure, the performance was actually quite good (Q3 = 85.8%, SOV = 86.5%). Comparisons to other predictors on the same set or proteins (PSIPRED, JNET) or a subset of these proteins (PORTER, PROF-KING, etc.) indicate that these methods perform at levels from 70.5%–77.1% (Q3) or 70.9%–77.9% (SOV). The Q3 and SOV scores we obtained for these predictors on our EVA test set are very close (<1% difference) to those reported by the authors or posted on the EVA website. While the performance of PROTEUS is not quite as impressive as seen in the first test, it still demonstrates that under strict "CASP" testing conditions, PROTEUS performs approximately 4–8% better than other high-performing secondary structure predictors.

Hisotgram comparing the Q3 (light) and SOV (dark) scores of PROTEUS (right 3 bars) versus PSIPRED, JNET, and TRANSSEC for the test set (N = 1644) of non-homologous EVA sequences. Data for YASPIN, PORTER, PROF-KING, PROFSEC, and SAM-T99 are also shown. These were calculated from a smaller (N = 30–39) subset of sequences and predictions posted on the EVA website and on the PROTEUS home page. The Q3 score is written at the top of each predictor's set of bars. Standard deviations are shown as error bars.
The third test of PROTEUS' performance was intended to simulate the situation where one is trying the predict the secondary structure of proteins that are being studied by X-ray and NMR, but not yet solved, not yet published or not yet released by the PDB. This kind of test is intended to answer the question: What is the secondary structure prediction performance of PROTEUS for proteins that are of interest to genome annotators, structural biologists or protein chemists? A testing set of 125 randomly chosen, non-redundant, water soluble proteins was generated by downloading the PDB coordinates of a subset of proteins deposited from January 2005 to June 2005. Because the training set of proteins originally used to refine and optimize PROTEUS consisted of proteins deposited into the PDB prior to December 2004, this precluded any possibility of testing on the training set. As with the previous tests, the secondary structure for each of the test-set proteins was assigned by VADAR [ 48 ]. PROTEUS was then used to predict the secondary structures (with the homologue search turned on or off) and the performance was evaluated against the VADAR-assigned secondary structures. Figure 3 summarizes the distribution of Q3 scores for PROTEUS as tested over the entire 125 protein test suite, with the homologue search turned off (i.e. using the de novo prediction only). The average score in this case was 79.7% (Q3) and 82.0% (SOV) with a standard deviation of 7.5% and 10.3%, respectively. Figure 4 displays the distribution of PROTEUS' Q3 scores with the homologue search turned as applied to the 88 proteins in the test set for which a PDB homologue (with an expect score >10 -7 ) was found. In other words, 70.4% of the test proteins could have their secondary structure predicted via 3D-to-2D mapping. The average score for the 88 homologues was 90.0% (Q3) and 91.8% (SOV) with a standard deviation of 6.3% and 7.0% for Q3 and SOV scores respectively. Therefore PROTEUS' combined, consensus prediction (Figure 5 ) for all 125 test proteins yielded an average accuracy of 87.8% and 90.0% for Q3 and SOV scores respectively, with a standard deviation of 7.9% (Q3) and 8.7% (SOV). The low scoring outlier proteins (Q3 scores between 62%–70%) are typically very short peptides or proteins which have absolutely no homologue in the PDB. For further comparison the same test set (125 proteins) and testing procedures were used to evaluate the performance of several other high-performing secondary structure prediction methods including PSIPRED [ 35 ], JNET [ 36 ], SAM_T02 [ 39 ], as well as a locally written version of GOR [ 53 ] and our own TRANSEC. To ensure complete consistency, the BLAST database searches, which were required for all programs (except GOR), were performed on the same local copy of the non-redundant (NR) NCBI protein database. Figure 6 presents the results of these prediction programs in comparison to the predictions obtained with PROTEUS. A quick visual comparison reveals that PROTEUS' performance is significantly better (10–30%) than all five tested programs. For instance, PSIPRED, which is generally regarded as being one of the most accurate methods [ 11 , 35 ], obtained scores of 78.1% (Q3) and 80.9% (SOV) respectively. In comparison, PROTEUS' consensus method obtained scores of 87.8% (Q3) and 90.0% (SOV). Therefore, in this test, PROTEUS' scores were approximately 10% higher than those achieved by PSIPRED. Even when PROTEUS is partially disabled (the PDB homologue search is turned off) it still performs about 2% better than the best-performing routine (79.7% vs. 78.1%). The statistical significance of this 2% improvement was verified using a standard paired two-sample t -test, which confirmed that the two means were statistically different (p = 4.63 × 10 -7 , t-stat = 5.166, critical value = 1.657 with 124 degrees of freedom).

Histogram illustrating the distribution of accuracy (Q3) scores (%) for PROTEUS' de novo secondary structure predictions (i.e. with the PDB homologue search turned off) as measured on the complete test set of 125 PDB entries. The mean is 79.7% and the standard deviation is 7.5%.

Histogram illustrating the distribution of accuracy (Q3) scores (%) as measured on the test set of 88 proteins that had homologs (E > 10 -7 ) to existing PDB entries. The mean is 90.0% and the standard deviation is 6.3%.

Histogram illustrating the distribution of accuracy (Q3) scores (%) of the consensus prediction from PROTEUS as measured on the test set of 125 proteins. The mean is 87.8% and the standard deviation is 10.2%.

Hisotgram comparing the Q3 (black) and SOV (gray) scores of PROTEUS (left 3 bars) versus PSIPRED, JNET, PHD, SAM-T02 and GOR for test set of 125 proteins. The Q3 score is written at the top of each predictor's set of bars. Standard deviations are shown as error bars.
To verify that the performance differences noted in Figure 5 were not the result of improper program installation, limited tool selection or outdated software, we conducted a fourth test on a set of 10 recently (Sept, 2005) solved proteins using a number of popular secondary structure prediction web servers. Note that these 10 proteins were not contained in the PROTEUS-2D database. The proteins ranged in size from 76–502 residues. The results are summarized in Table 1 . Once again, the results largely confirm what was seen in Figure 5 , with PROTEUS averaging close to 90% in both Q3 and SOV and the others ranging between 55% and 75%. The performance of these servers in this test set is also consistent with what has been described in the literature [ 11 , 35 – 39 ]. Overall, these four independent tests confirm that PROTEUS is able to predict secondary structure of soluble proteins with very high accuracy. When restricted to the prediction of sequence-unique proteins (such as those found in EVA or those targets selected for structural genomics projects) PROTEUS has a Q3 of 81.3%, which as about 4–8% better than the best performing methods. When allowed to predict the structure of any generic protein (as might be done for a genome annotation project) PROTEUS has a Q3 of 88%–90% which is about 12–15% better than the best performing methods described to date.
PROTEUS was primarily developed to facilitate secondary structure prediction for genome annotation. In genome annotation one is primarily interested in getting the most correct annotations or the most accurate predictions in the quickest possible way. Making use of prior information or fragmentary data to fill in knowledge gaps is perfectly reasonable and strongly encouraged [ 16 , 14 , 21 , 22 , 29 ]. Likewise making this process as automated and fool-proof as possible is a basic requirement of genome annotation systems. If one is interested in getting the most complete and accurate secondary structure assignment of as many proteins as possible, then it is quite natural to want to combine an ab initio or de novo prediction method with a method that extracts known or partially known secondary structure assignments (from PDB data, from NMR NOE data, from MS/MS hydrogen exchange data) and to have this done automatically.
Perhaps the best way to appreciate the general utility of PROTEUS is to imagine a scenario where one is given the sequence of a large 840 residue protein (lets call it Vav1) and then asked to generate the most accurate or most correct secondary structure assignment for this protein. Suppose a BLAST search or CDD search reveals that this protein has 7 different domains, 4 of which have PDB homologues (2 of which have less than 35% sequence identity to a PDB target) and 3 other domains which have no known structure. To generate the most accurate possible secondary structure assignment for this multidomain protein would require many manual steps and a good deal of bioinformatics skill including: 1) a BLAST search against the PDB; 2) manual selection of the highest scoring homologues; 3) homology modeling using Swiss-Model [ 42 ] or another modeling server for the two homologous domains with >35% sequence identity; 4) assignment of the secondary structure for two of the domains using DSSP, STRIDE or VADAR; 5) sequence-based threading on the 3D-PSSM server [ 28 ] to generate possible folds of the remaining two low-scoring homologues; 6) manual assessment and adjustment of the predicted folds and their alignments; 7) prediction of the secondary structure of the remaining 3 domains using a de novo predictor such as PSIPRED or PHD and 8) manually typing, cutting or pasting all the secondary structure assignments on to every residue in the 840 residue sequence. A skilled bioinformatician might be able to do this in a couple of hours, an unskilled individual might take several days. Alternately, one may elect the easy route and simply predict the structure of the entire protein using a de novo structure predictor such as PSIPRED or PHD. However, choosing to do this would likely reduce the accuracy of the prediction by 10–15% (i.e. going from a Q3 of 85% to 75%).
Now suppose that one was asked to do this kind of high-end structure prediction not for just one protein but for 23,000 proteins (i.e. genome annotation) or that it has to be done on 4000 proteins every 2 weeks (the current rate at which new microbial genomes are being released). Clearly such a manual intensive process would have to be replaced by an automated technique. This is the primary motivation behind PROTEUS. PROTEUS effectively replaces 8 manually tedious steps with a single automated process. In fact, this 8 step example of Vav1 is not entirely hypothetical. The single step PROTEUS result (which takes about 2 minutes) for Vav1 is shown in the Sample Output on the PROTEUS homepage. Inspection of the output clearly demonstrates how PROTEUS can combine prior knowledge (PDB data) with de novo predictions to generate optimally accurate secondary structure assignments for large and complex proteins.
PROTEUS is able to achieve its very high level of accuracy because it brings together two high performing methods of secondary structure prediction – a novel de novo method based on a jury-of-experts approach and a novel 3D-to-2D homology mapping method. The 3D-to-2D mapping process is not completely unknown. In fact, it is frequently used as an intermediate step in several homology modeling programs to identify conserved structural scaffolds [ 7 , 42 ]. Given the well known fact that secondary structure is more conserved than primary structure, it stands to reason that mapping the secondary structure onto a given query sequence – even for remotely related homologues – will yield a high quality secondary structure "prediction". This is borne out by the fact that our mapping method is able to predict secondary structure with greater than 90% accuracy. This mapping approach is obviously limited to query proteins that have a homologue or potential homologue already deposited in the PDB database. As might be expected, the accuracy of the mapping prediction is generally tied to the level of sequence identity or BLAST expect value. Highly similar sequences (>80% identity) can have their secondary structure predicted with close to 90% accuracy. Intermediate similarity (40–80% identity) yields predictions that are 80–90% correct while low sequence identity (25–40%) yields secondary structure predictions that are 75–80% correct. This partly explains the distribution of scores seen in Figure 4 .
Certainly, when the PDB was relatively small (prior to the year 2000), this 3D-to-2D mapping method would prove to be relatively ineffective. However, with the rapid expansion of the PDB over the past 5 years we are now able to take advantage of the fact that an increasingly large fraction of protein structures that are being solved or for which people want to know the structure, have at least one homologue in the Protein Data Bank. Indeed, less than 3% of all newly deposited structures have novel folds (and therefore novel secondary structure arrangements) and it appears that less than 10% of structural genomics targets are yielding truly novel folds [ 43 – 45 ]. Therefore, the odds that any given protein will have a novel arrangement or a unique order of secondary structures (which would reduce the accuracy of this homologue approach) is becoming relatively small. Even with the modest approach employed here (requiring sequence identity >25% or an E < 10 -7 ), we still find that 70% of "testable" proteins have at least one homologue or a portion of a homologue in the PDB. Therefore, on average, the 3D-to-2D mapping process is going to be effective for about 70% of all query proteins which are solvable by today's X-ray and NMR methods. We would predict that this fraction (70%) would continue to increase as the PDB continues to expand and the number of known folds grows.
Note that this figure of 70% is not applicable if were to try to predict secondary structure for entire genomes. Large scale homology modeling efforts suggest that only about 30–50% of a given genome is amenable to homology modeling or threading [ 54 ]. Therefore if we applied the lower figure of 30% (for the probability of finding a PDB homologue in a newly sequenced genome) to our protocol we would predict the performance of PROTEUS in predicting the secondary structure of soluble proteins would drop to 83%. Note that this figure is still 7–10% better than existing secondary structure prediction methods. Obviously if one biased their selection of query proteins such that no portion of the sequence had any sequence homology whatsoever to something in the PDB, then PROTEUS could do no better than its de novo approach (about 78–79%), even with its PDB search turned on. Similarly, we would predict that genomes from poorly sampled branches of the tree-of-life would probably be less well predicted than those belonging to the better studied branches (mouse, yeast, humans, E. coli).
Given the potential variability in PROTEUS' predictions, we believed it was important to provide a reliability or confidence score in PROTEUS' prediction output. These reliability scores are determined on the basis of the neural network outputs (for the de novo predictions) or the level of sequence identity to a given PDB match (for the 3D-to-2D mapping method). Reliability scores are generated not only for each residue for each prediction, but also for each residue in the consensus (i.e. final) prediction and for the entire protein. The maximum reliability score is 9 (for a residue) and the maximum reliability score for a complete protein is 90%.
While PROTEUS' 3D-to-2D mapping procedure offers a number of advantages in secondary structure prediction, it is also important to remember that another key strength in PROTEUS lies in its de novo structure prediction routine. This jury-of-experts approach, which uses machine learning methods to combine three independent and high performing structure prediction algorithms into one, is able to consistently predict secondary structures with an accuracy approaching 79%. This is still 2% higher than any other single pass method with which we could directly compare. This consensus method uses PSIPRED, which generates BLAST sequence profiles to extract evolutionary and sequence information using a neural network; JNET, which uses a combination of solubility information, evolutionary information, and a Hidden-Markov Model/neural network combination; and TRANSSEC, a locally developed algorithm which uses a two-tiered prediction system to extract evolutionary similarities. These three methods are sufficiently "orthogonal" in their prediction methodology that the combination of the three is able to generate a consensus prediction that is 2–5% higher than any individual prediction. The ability to generate de novo secondary structure predictions which are consistently near 80% correct, especially in regions where the 3D-to-2D mapping approach fails, certainly helps to create consensus predictions that are consistently close to 88% correct.
While PROTEUS clearly performs very well, there are still a number of improvements or additions that could be made to the program. One obvious improvement could be the integration of conventional membrane spanning prediction routines and signal recognition programs [ 55 ] to make PROTEUS capable of handling all protein types (water-soluble, targeted and transmembrane proteins). This would be particularly useful in whole genome annotation applications. Another improvement could be made in PROTEUS' sensitivity in its 3D-to-2D mapping steps. By simply employing PSI-BLAST [ 56 ] instead of BLAST it should be possible to increase the fraction of PDB homologues (from 70% to ~80%) that could pass through the 3D-to-2D mapping steps. However, given the drop in predictive performance seen for homologues with <30% identity, it is not clear whether this would lead to a very substantial improvement in overall accuracy. Yet another potential addition to PROTEUS would be a 2D threading or fold prediction service. Given the high accuracy of its secondary structure predictions, one might expect that PROTEUS could yield somewhat more reliable results and somewhat improved fold classifications.
Along with its high accuracy and its ready availability as a web server, we have also ensured that the downloadable version of PROTEUS would be a well-documented, user-friendly system which is easy to install and does not require additional input or obscure pre-processing steps. During our testing processes we found that many other systems offered relatively limited documentation, required the user to provide additional inputs, such as an alignment and BLAST output files, or demanded that additional scripts or programs to be run to compile the input into a suitable format. Often users will not know how to supply these extra inputs (for example, creating a list of aligned sequences in a special format). Given these difficulties, we have tried to make the installation and operation of PROTEUS as simple as possible. The local version of PROTEUS (see Availability and Requirements section) requires nothing more than a sequence in either FASTA or Raw format. The output can be customized, and due to its open source nature, modular design and extensively commented Java code, the algorithms can be incorporated easily into other applications for batch or online processing. PROTEUS was also designed to take full advantage of multi-processor systems and should scale well as computational resources increase. This is a particularly important consideration in genome/proteome annotation efforts.
PROTEUS' software does have a few drawbacks. Because it is written in Java, it requires substantial memory to run. Furthermore, the neural networks used in the program were not optimized for minimal memory use; therefore PROTEUS requires at least 512 MB of RAM to be allocated to the Java Virtual Machine. With increasing hardware availability and lower prices, this requirement should not be too much of a concern in the future. Additionally, because of the requirement to run three independent de novo prediction methods, a 3D-to-2D mapping step and a consensus prediction generator, PROTEUS is somewhat slower than other methods. While PSIPRED can typically return a result within seconds of completing a lengthy PSI-BLAST search, PROTEUS requires almost a minute to complete its predictions (in addition to a PSIBLAST search). Efforts are being made to reduce this time requirement with further code optimization and multi-processor utilization.
PROTEUS is both an integrated web server and a stand-alone application that exploits recent advancements in data mining and machine learning to perform very accurate protein secondary structure predictions. PROTEUS combines three high-performing de novo structure prediction methods (PSIPRED, JNET and TRANSSEC), a jury-of-experts consensus tool and a robust PDB-based structure alignment process to generate all of its secondary structure predictions. For water-soluble protein PROTEUS is able to achieve a very high level of accuracy (Q3 = 88%, SOV = 90%) which is approximately 12–15% higher than that previously reported [ 35 – 41 ]. The program's performance was extensively tested and compared to both available programs and publicly accessible web servers using a variety of test proteins and test scenarios. In all cases PROTEUS appears to perform better than existing tools. This performance improvement is statistically significant and robust. In the rare situations (20–30%) where a query protein shows no similarity whatsoever to any known structure, or if the 3D-to-2D mapping option is turned off, PROTEUS is still able to achieve a Q3 score of ~79%. This is still statistically better than what has been reported elsewhere. However, it is still important to be somewhat circumspect in interpreting these results. The standard deviation for essentially all secondary structure prediction routines (including PROTEUS) still stands at ~10% and so some caution must be exercised in interpreting or relying upon these predictions. Indeed, it is theoretically possible to get a PROTEUS prediction that is only 50% correct. Until a method is developed where the standard deviation in prediction accuracy is <5% or until the PDB expands to encompass all "fold space", there is still a strong need to develop better routines and more complete databases. To facilitate further algorithmic improvements, widespread adoption, and easy incorporation into genome annotation pipelines, PROTEUS was designed to be completely open source. Given its high accuracy and open-source nature, we believe PROTEUS could make a very useful addition to the current arsenal of structure prediction tools available to protein chemists, genome annotators and bioinformaticians.
Availability and requirements
The PROTEUS website is accessible at http://wishart.biology.ualberta.ca/proteus . The entire PROTEUS suite occupies approximately 1.2 GBytes of data with the PROTEUS-2D database occupying 5.2 Mbytes and the NR protein sequence database occupying 1.1 Gbytes. All programs were tested and compiled on a variety of UNIX platforms and should work on most systems operating Linux and Mac OSX (10.4+). All programs and databases are downloadable at http://129.128.185.184/proteus/contact.jsp and are supported with an easy-to-use installation script. A typical PROTEUS run for a 300 residue sequence takes approximately 3 minutes on a 2.8 GHz machine equipped with 1 GB of RAM.
Pauling L, Corey RB, Branson HR: The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci USA 1951, 37: 205–234. 10.1073/pnas.37.4.205
Article PubMed Central CAS PubMed Google Scholar
Guzzo AV: The influence of amino acid sequence on protein structure. Biophys J 1965, 5: 809–822.
Chou PY, Fasman GD: Prediction of protein conformation. Biochemistry 1974, 13: 222–245. 10.1021/bi00699a002
Article CAS PubMed Google Scholar
Westbrook JD, Feng Z, Chen L, Yang H, Berman HM: The Protein Data Bank and structural genomics. Nucleic Acids Res 2003, 31: 489–491. 10.1093/nar/gkg068
Engelman DM, Steitz TA, Goldman A: Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu Rev Biophys Biophys Chem 1986, 15: 321–353. 10.1146/annurev.bb.15.060186.001541
Jones DT, Taylor WR, Thornton JM: A new approach to protein fold recognition. Nature 1992, 358: 86–89. 10.1038/358086a0
Sutcliffe MJ, Haneef I, Carney D, Blundell TL: Knowledge based modelling of homologous proteins, Part I: Three-dimensional frameworks derived from the simultaneous superposition of multiple structures. Protein Eng 1987, 1: 377–384.
Rost B, Schneider R, Sander C: Protein fold recognition by prediction-based threading. J Mol Biol 1997, 270: 471–480. 10.1006/jmbi.1997.1101
Rost B: Review: protein secondary structure prediction continues to rise. J Struct Biol 2001, 134: 204–218. 10.1006/jsbi.2001.4336
Lattman EE: Fifth Meeting on the Critical Assessment of Techniques for Protein Structure Prediction. Proteins 2003, 53 (Suppl 6):33.
Google Scholar
Eyrich VA, Marti-Renom MA, Przybylski D, Madhusudhan MS, Fiser A, Pazos F, Valencia A, Sali A, Rost B: EVA: continuous automatic evaluation of protein structure prediction servers. Bioinformatics 2001, 17: 1242–1243. 10.1093/bioinformatics/17.12.1242
Cozzetto D, Di Matteo A, Tramontano A: Ten years of predictions ... and counting. FEBS J 2005, 272: 881–882.
Rost B, Yachdav G, Liu J: The PredictProtein server. Nucleic Acids Res 2004, (32 Web Server):W321–326.
Van Domselaar GH, Stothard P, Shrivastava S, Cruz JA, Guo A, Dong X, Lu P, Szafron D, Greiner R, Wishart DS: BASys: a web server for automated bacterial genome annotation. Nucleic Acids Res 2005, (33 Web Server):W455–459. 10.1093/nar/gki593
Gardy JL, Spencer C, Wang K, Ester M, Tusnady GE, Simon I, Hua S, deFays K, Lambert C, Nakai K, Brinkman FS: PSORT-B: Improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res 2003, 31: 3613–3617. 10.1093/nar/gkg602
Mewes HW, Frishman D, Mayer KF, Munsterkotter M, Noubibou O, Pagel P, Rattei T, Oesterheld M, Ruepp A, Stumpflen V: MIPS: analysis and annotation of proteins from whole genomes in 2005. Nucleic Acids Res 2006, (34 Database):D169–172. 10.1093/nar/gkj148
Stothard P, Van Domselaar G, Shrivastava S, Guo A, O'Neill B, Cruz J, Ellison M, Wishart DS: BacMap: an interactive picture atlas of annotated bacterial genomes. Nucleic Acids Res 2005, (33 Database):D317–320.
Gibbs AC, Bjorndahl TC, Hodges RS, Wishart DS: Probing the structural determinants of type II' beta-turn formation in peptides and proteins. J Am Chem Soc 2002, 124: 1203–1213. 10.1021/ja011005e
Ullman CG, Haris PI, Smith KF, Sim RB, Emery VC, Perkins SJ: Beta-sheet secondary structure of an LDL receptor domain from complement factor I by consensus structure predictions and spectroscopy. FEBS Lett 1995, 371: 199–203. 10.1016/0014-5793(95)00916-W
Lee S, Cho MK, Jung JW, Kim JH, Lee W: Exploring protein fold space by secondary structure prediction using data distribution method on Grid platform. Bioinformatics 2004, 20: 3500–3507. 10.1093/bioinformatics/bth435
Carter P, Liu J, Rost B: PEP: Predictions for Entire Proteomes. Nucleic Acids Res 2003, 31: 410–413. 10.1093/nar/gkg102
Liu J, Rost B: Comparing function and structure between entire proteomes. Protein Sci 2001, 10: 1970–1979. 10.1110/ps.10101
Wishart DS, Case DA: Use of chemical shifts in macromolecular structure determination. Methods Enzymol 2001, 338: 3–34.
Wang Y, Wishart DS: A simple method to adjust inconsistently referenced 13C and 15N chemical shift assignments of proteins. J Biomol NMR 2005, 31: 143–148. 10.1007/s10858-004-7441-3
Vainshtein I, Atrazhev A, Eom SH, Elliott JF, Wishart DS, Malcolm BA: Peptide rescue of an N-terminal truncation of the Stoffel fragment of taq DNA polymerase. Protein Sci 1996, 5: 1785–1792.
Grasselli E, Noviello G, Rando C, Nicolini C, Vergani L: Expression, purification and characterisation of a novel mutant of the human protein kinase CK2. Mol Biol Rep 2003, 30: 97–106. 10.1023/A:1023934805326
Szafron D, Lu P, Greiner R, Wishart DS, Poulin B, Eisner R, Lu Z, Anvik J, Macdonell C, Fyshe A, Meeuwis D: Proteome Analyst: custom predictions with explanations in a web-based tool for high-throughput proteome annotations. Nucleic Acids Res 2004, (32 Web Server):W365–371.
Kelley LA, MacCallum RM, Sternberg MJ: Enhanced genome annotation using structural profiles in the program 3D-PSSM. J Mol Biol 2000, 299: 499–520. 10.1006/jmbi.2000.3741
Clare A, Karwath A, King RD: Functional bioinformatics for Arabidopsis thaliana. Bioinformatics , in press. 2006, Feb 15 2006, Feb 15
Lin K, Simossis VA, Taylor WR, Heringa J: A simple and fast secondary structure prediction method using hidden neural networks. Bioinformatics 2005, 21: 152–159. 10.1093/bioinformatics/bth487
Pollastri G, McLysaght A: Porter: a new, accurate server for protein secondary structure prediction. Bioinformatics 2005, 21: 1719–1720. 10.1093/bioinformatics/bti203
Ouali M, King RD: Cascaded multiple classifiers for secondary structure prediction. Protein Sci 2000, 9: 1162–1176.
Karplus K, Karchin R, Barrett C, Tu S, Cline M, Diekhans M, Grate L, Casper J, Hughey R: What is the value added by human intervention in protein structure prediction? Proteins 2001, (Suppl 5):86–91. 10.1002/prot.10021
Rost B, Eyrich VA: EVA: large-scale analysis of secondary structure prediction. Proteins 2001, (Suppl 5):192–199. 10.1002/prot.10051
Jones DT: Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 1999, 292: 195–202. 10.1006/jmbi.1999.3091
Cuff JA, Barton GJ: Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins 2000, 40: 502–511. 10.1002/1097-0134(20000815)40:3<502::AID-PROT170>3.0.CO;2-Q
Rost B, Sander C, Schneider R: PHD – an automatic mail server for protein secondary structure prediction. Comput Appl Biosci 1994, 10: 53–60.
CAS PubMed Google Scholar
Rost B: PHD: predicting one-dimensional protein structure by profile based neural networks. Meth Enzymol 1996, 266: 525–539.
Karplus K, Karchin R, Draper J, Casper J, Mandel-Gutfreund Y, Diekhans M, Hughey R: Combining local-structure, fold-recognition, and new fold methods for protein structure prediction. Proteins 2003, 53 (Suppl 6):491–496. 10.1002/prot.10540
Ginalski K, Elofsson A, Fischer D, Rychlewski L: 3D-Jury: A simple approach to improve protein structure predictions. Bioinformatics 2003, 19: 1015–1018. 10.1093/bioinformatics/btg124
Eyrich VA, Rost B: META-PP: single interface to crucial prediction servers. Nucleic Acids Res 2003, 31: 3308–3310. 10.1093/nar/gkg572
Schwede T, Kopp J, Guex N, Peitsch MC: SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res 2003, 31: 3381–3385. 10.1093/nar/gkg520
Amegbey GY, Stothard P, Kuznetsova E, Yee A, Arrowsmith CH, Wishart DS: Solution structure of MTH0776 from methanobacterium thermoautotrophicum. J Biomol NMR , in press.
McGuffin LJ, Jones DT: Targeting novel folds for structural genomics. Proteins 2002, 48: 44–52. 10.1002/prot.10129
Yee A, Pardee K, Christendat D, Savchenko A, Edwards AM, Arrowsmith CH: Structural proteomics: toward high-throughput structural biology as a tool in functional genomics. Acc Chem Res 2003, 36: 183–189. 10.1021/ar010126g
Wishart DS, Boyko RF, Sykes BD: Constrained multiple sequence alignment using XALIGN. Comput Appl Biosci 1994, 10: 687–688.
Li W, Jaroszewski L, Godzik A: Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics 2001, 17: 282–283. 10.1093/bioinformatics/17.3.282
Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, Wishart DS: VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res 2003, 31: 3316–3319. 10.1093/nar/gkg565
Kabsch W, Sander C: Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22: 2577–2637. 10.1002/bip.360221211
Heinig M, Frishman D: STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res 2004, (32 Web Server):W500–502.
Jooneworld resources [ http://www.jooneworld.com ]
Zemla A, Venclovas C, Fidelis K, Rost B: A modified definition of Sov, a segment-based measure for protein secondary structure prediction assessment. Proteins 1999, 34: 220–223. 10.1002/(SICI)1097-0134(19990201)34:2<220::AID-PROT7>3.0.CO;2-K
Garnier J, Osguthorpe DJ, Robson B: Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J Mol Biol 1978, 120: 97–120. 10.1016/0022-2836(78)90297-8
Pieper U, Eswar N, Stuart AC, Ilyin VA, Sali A: MODBASE, a database of annotated comparative protein structure models. Nucleic Acids Res 2002, 30: 255–259. 10.1093/nar/30.1.255
Kernytsky A, Rost B: Static benchmarking of membrane helix predictions. Nucleic Acids Res 2003, 31: 3642–3654. 10.1093/nar/gkg532
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25: 3389–3402. 10.1093/nar/25.17.3389
HNN [ http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_nn.html ]
JPRED [ http://www.compbio.dundee.ac.uk/~www-jpred/ ]
NNPredict [ http://www.cmpharm.ucsf.edu/~nomi/nnpredict.html ]
SSPro [ http://www.igb.uci.edu/tools/scratch/ ]
Porter [ http://distill.ucd.ie/porter/ ]
Sopma [ http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html ]
Prof [ http://www.aber.ac.uk/~phiwww/prof/ ]
Download references
Acknowledgements
Funding for this project was provided by the Protein Engineering Network of Centres of Excellence (PENCE), NSERC and Genome Prairie (a division of Genome Canada).
Author information
Authors and affiliations.
Department of Computing Science, University of Alberta, Edmonton, AB, T6G 2E8, Canada
Scott Montgomerie, Shan Sundararaj, Warren J Gallin & David S Wishart
Department of Biological Sciences, University of Alberta, Edmonton, AB, T6G 2E9, Canada
David S Wishart
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to David S Wishart .
Additional information
Authors' contributions.
SM wrote, tested and installed most of the predictive software described here, designed and conducted all performance tests and prepared the first draft of the manuscript, SS wrote and tested the software used to generate the PROTEUS-2D database, WJG provided direction, ideas and critical suggestions in the early phases of the project, DSW wrote the final manuscript, conceived of the central ideas in the paper and coordinated most of the project.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Authors’ original file for figure 1
Authors’ original file for figure 2, authors’ original file for figure 3, authors’ original file for figure 4, authors’ original file for figure 5, authors’ original file for figure 6, rights and permissions.
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Reprints and permissions
About this article
Cite this article.
Montgomerie, S., Sundararaj, S., Gallin, W.J. et al. Improving the accuracy of protein secondary structure prediction using structural alignment. BMC Bioinformatics 7 , 301 (2006). https://doi.org/10.1186/1471-2105-7-301
Download citation
Received : 18 October 2005
Accepted : 14 June 2006
Published : 14 June 2006
DOI : https://doi.org/10.1186/1471-2105-7-301
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Secondary Structure
- Protein Data Bank
- Query Sequence
- Confidence Score
- Secondary Structure Prediction
BMC Bioinformatics
ISSN: 1471-2105
- General enquiries: [email protected]
- PRABI-GERLAND RHONE-ALPES BIOINFORMATIC POLE GERLAND SITE
- Institute of Biology and Protein Chemistry
- Publications

An improved multi-scale convolutional neural network with gated recurrent neural network model for protein secondary structure prediction
- Original Article
- Published: 13 May 2024
Cite this article

- Vrushali Bongirwar 1 &
- A. S. Mokhade 2
75 Accesses
Explore all metrics
Protein structure prediction is one of the main research areas in the field of Bio-informatics. The importance of proteins in drug design attracts researchers for finding the accurate tertiary structure of the protein which is dependent on its secondary structure. In this paper, we focus on improving the accuracy of protein secondary structure prediction. To do so, a Multi-scale convolutional neural network with a Gated recurrent neural network (MCNN-GRNN) is proposed. The novel amino acid encoding method along with layered convolutional neural network and Gated recurrent neural network blocks helps to retrieve local and global relationships between features, which in turn effectively classify the input protein sequence into 3 and 8 states. We have evaluated our algorithm on CullPDB, CB513, PDB25, CASP10, CASP11, CASP12, CASP13, and CASP14 datasets. We have compared our algorithm with different state-of-the-art algorithms like DCNN-SS, DCRNN, MUFOLD-SS, DLBLS_SS, and CGAN-PSSP. The Q3 accuracy of the proposed algorithm is 82–87% and Q8 accuracy is 69–77% on different datasets.
This is a preview of subscription content, log in via an institution to check access.
Access this article
Price excludes VAT (USA) Tax calculation will be finalised during checkout.
Instant access to the full article PDF.
Rent this article via DeepDyve
Institutional subscriptions
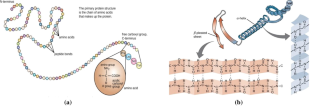
Similar content being viewed by others

Prediction of 8-state protein secondary structures by a novel deep learning architecture

A Comparative Study of Various Deep Learning Architectures for 8-state Protein Secondary Structures Prediction

Deep Learning-Based Experimentation for Predicting Secondary Structure of Amino Acid Sequence
Data availability.
Data used in this study is publicly available.
VrushaliBongirwar ASM (2022) Different methods, techniques and their limitations in protein structure prediction: a review. Progr Biophys Mol Biol 173:72–82. https://doi.org/10.1016/j.pbiomolbio.2022.05.002
Article Google Scholar
Chou K-C (2001) Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 43(3):246–255. https://doi.org/10.1002/prot.10355
Gao CF, Wu XY (2018) Feature extraction method for proteins based on markov tripeptide by compressive sensing. BMC Bioinform 19(1):239–232. https://doi.org/10.1186/s12859-018-2235-x
Jones DT (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292(2):195–202. https://doi.org/10.1006/jmbi.1999.3091
Smolarczyk T, Roterman-Konieczna I, Stapor K (2020) Protein secondary structure prediction: a review of progress and directions. Curr Bioinform 15:90–107. https://doi.org/10.2174/1574893614666191017104639
Chou PY, Fasman GD (1974) Conformational parameters for amino acids in helical, B-sheet, and random coil regions calculated from proteins. Biochemistry 13(2):211–222
Rost B, Sander C (1993) Prediction of protein secondary structure at better than 70% accuracy. J Mol Biol 232(2):584–599. https://doi.org/10.1006/jmbi.1993.1413
McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16(4):404–405. https://doi.org/10.1093/bioinformatics/16.4.404
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Ward JJ, McGuffin LJ, Buxton BF, Jones DT (2003) Secondary structure prediction with support vector machines. Bioinformatics 19(3):1950–1955. https://doi.org/10.1093/bioinformatics/btg223
Dor O, Zhou Y (2007) Achieving 80% ten-fold cross-validated accuracy for secondary structure prediction by large-scale training. Proteins 66(4):838–845. https://doi.org/10.1002/prot.21298
Faraggi E, Zhang T, Yang Y, Lukasz Kurgan YZ (2011) SPINE X: improving protein secondary structure prediction by multistep learning coupled with prediction of solvent accessible surface area and backbone torsion angles. J Comput Chem 33(3):259–267. https://doi.org/10.1002/jcc.21968
Bettella F, Rasinski D, Knapp EW (2012) Protein secondary structure prediction with SPARROW. J Chem Inf Model 52(2):545–556. https://doi.org/10.1021/ci200321u
Ashraf Yaseen YL (2014) Context-based features enhance protein secondary structure prediction accuracy. J Chem Inf Model 54(3):992–1002. https://doi.org/10.1021/ci400647u
Drozdetskiy A, Cole C, Procter J, Barton GJ (2015) JPred4: a protein secondary structure prediction server. Nucleic Acids Res 43(April):389–394. https://doi.org/10.1093/nar/gkv332
Heffernan R, Paliwal K, Lyons J, Dehzangi A, Sharma A (2015) Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Nature Publishing Group. https://doi.org/10.1038/srep11476
Book Google Scholar
Wang S, Peng J, Ma J, Xu J (2016) Protein secondary structure prediction using deep convolutional neural fields. Sci Rep. https://doi.org/10.1038/srep18962
Heffernan R, Yang Y, KuldipPaliwal YZ (2017) Capturing non-local interactions by long short-term memory bidirectional recurrent neural networks for improving prediction of protein secondary structure, backbone angles, contact numbers and solvent accessibility. Bioinformatics 33(18):2842–2849. https://doi.org/10.1093/bioinformatics/btx218
Zhen Li YY (2016). Protein secondary structure prediction using cascaded convolutional and recurrent neural networks. In: Proceedings of the Twenty-Fifth International Joint Conference on Artificial Intelligence (IJCAI-16), 2560–2567.
Fang C, Shang Y, Xu D (2018) MUFOLD-SS: new deep inceptioninside- inception networks for protein secondary structure prediction. Proteins 86(5):592–598. https://doi.org/10.1002/prot.25487
Guo L, Jiang Q, Jin X, Liu L, Zhou W, Yao S (2020) A deep convolutional neural network to improve the prediction of protein secondary structure. Curr Bioinform 15:767–777. https://doi.org/10.2174/1574893615666200120103050
Yuan L, Hu X, Ma Y, Liu Y (2022) DLBLS_SS: protein secondary structure prediction using deep learning and broad learning system. RSC Adv 12:33479–33487. https://doi.org/10.1039/d2ra06433b
Jin X, Guo L, Jiang Q, Wu N (2022) Prediction of protein secondary structure based on an improved channel attention and multiscale convolution module. Front Bioeng Biotechnol 10:901018. https://doi.org/10.3389/fbioe.2022.901018
Hasic H, Buza E, Akagic A (2017) A hybrid method for prediction of protein secondary structure based on multiple artificial neural networks. In: 2017 40th international convention on information and communication technology, electronics and microelectronics (MIPRO) pp. 1195-1200. IEEE. https://doi.org/10.23919/MIPRO.2017.7973605
Yavuz BÇ, Yurtay N, Ozkan O (2018) Prediction of protein secondary structure with clonal selection algorithm and multilayer perceptron. IEEE Access 6:45256–45261. https://doi.org/10.1109/ACCESS.2018.2864665
Pollastri G, Przybylski D, Rost B (2003) Improving the prediction of protein secondary structure in three and eight classes using recurrent neural networks and profiles. Proteins 47(2):228–235. https://doi.org/10.1002/prot.10082
Wang Z, Zhao F, Peng J, Xu J (2011) Protein 8-class secondary structure prediction using conditional neural fields. Proteomics 11(19):3786–3792. https://doi.org/10.1002/pmic.201100196
Busia A, Jaitly N (2017). Next-step conditioned deep convolutional neural networks to improve protein secondary structure prediction. In: Conference on intelligent systems for molecular biology & European conference on computational biology, 1–11. https://doi.org/10.48550/arXiv.1702.03865
Zhou JTO (2014). Deep supervised and convolutional generative stochastic network for protein secondary structure prediction. In: Proceedings of the 31st International Conference on International Conference on Machine Learning Beijing, China, 1–9. https://doi.org/10.48550/arXiv.1403.1347
Fang C, Shang Y, Xu D (2017). A new deep neighbor residual network for protein secondary structure prediction. In: IEEE 29th International Conference on Tools with Artificial Intelligence (ICTAI), Boston, MA, USA, 66–71.
Guo Y, Li W, Wang B, Liu H, Zhou D (2019) DeepACLSTM: deep asymmetric convolutional long short-term memory neural models for protein secondary structure prediction. BMC Bioinform 20(341):1–12. https://doi.org/10.1186/s12859-019-2940-0
Drori I, Dwivedi I, Shrestha P, Wan J, Wang Y, He Y, Mazza A, Krogh-Freeman H, Leggas D, Sandridge K, Nan L, Thakoor K, Joshi C, Goenka S, Keasar C, (2018). High quality prediction of protein q8 secondary structure by diverse neural network architectures. NIPS 2018 Workshop on Machine Learning for Molecules and Materials, 1–10. https://doi.org/10.48550/arXiv.1811.07143
Yang W, Hu Z, Zhou L, Jin Y (2022) Knowledge-based systems protein secondary structure prediction using a lightweight convolutional network and label distribution aware margin loss. Knowl-Based Syst 237:1–12. https://doi.org/10.1016/j.knosys.2021.107771
Hinton GE, Krizhevsky A, Srivastava N, Sutskever I, Salakhutdinov R (2014) Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res 15:1929–1958
MathSciNet Google Scholar
Download references
Acknowledgements
We thank the Department of Computer Science and Engineering, Shri Ramdeobaba College of Engineering and Management, Nagpur for providing NVIDIA GPU A100 for experimentation.
Authors state no funding is involved.
Author information
Authors and affiliations.
Department of Computer Science and Engineering, Shri Ramdeobaba College of Engineering and Management, Nagpur, India
Vrushali Bongirwar
Department of Computer Science and Engineering, Visvesvaraya National Institute of Technology, Nagpur, India
A. S. Mokhade
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Vrushali Bongirwar .
Ethics declarations
Conflict of interest.
The authors declare that they have no relevant financial or non-financial/ competing interests to disclose in any material discussed in this article.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Reprints and permissions
About this article
Bongirwar, V., Mokhade, A.S. An improved multi-scale convolutional neural network with gated recurrent neural network model for protein secondary structure prediction. Neural Comput & Applic (2024). https://doi.org/10.1007/s00521-024-09822-8
Download citation
Received : 24 August 2023
Accepted : 15 April 2024
Published : 13 May 2024
DOI : https://doi.org/10.1007/s00521-024-09822-8
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Protein secondary structure prediction
- Multi-scale convolutional neural network
- Gated recurrent neural network
- Amino acid sequence encoding
- Find a journal
- Publish with us
- Track your research
IMAGES
VIDEO
COMMENTS
PHD SECONDARY STRUCTURE PREDICTION METHOD. [ Abstract ] [ NPS@ help ] [ Original server ] Sequence name (optional) : Paste a protein sequence below : help. Output width : User : public Last modification time : Mon Mar 15 15:24:33 2021. Current time : Sun May 12 03:21:50 2024.
PHD - neural network algorithm for secondary structure prediction Rost and Sander • First step - multiple alignment (say for the sequence family recovered by BLAST) • PHD uses two levels of Neural Networks • Level 1: Sequence to structure network: feed forward NN with 3 layers - input, hidden, output; responsible
Lectures as a part of various bioinformatics courses at Stockholm University
Early secondary structure prediction methods (such as Chou-Fasman and GOR, out-lined below) had a 3-state accuracy of 50{60%. (They initially reported higher accu- ... before moving on to one of the most successful recent methods, PhD, which is based on neural nets. 1It is possible to have more detailed descriptions ofsecondary structure ...
The prediction methods include Chou-Fasman, Garnier, Osguthorpe and Robson (GOR), PHD, neural network (NN)-based protein secondary structure prediction (PSIPRED), SPINE-X, protein secondary structure prediction (PSSpred) and meta methods. The chapter assesses the performance of different methods using the Q 3 measure. It investigates the ...
When he saw CASP13 results, Konstantin Weissenow, now a PhD student in the Rost lab, was a master's degree student working on a protein structure prediction method.
Abstract. Protein Structure Prediction is a central topic in Structural Bioinformatics. Since the '60s statistical methods, followed by increasingly complex Machine Learning and recently Deep Learning methods, have been employed to predict protein structural information at various levels of detail. In this review, we briefly introduce the ...
Protein secondary structure prediction (PSSP) is a fundamental task in protein science and computational biology, and it can be used to understand protein 3-dimensional (3-D) structures, further, to learn their biological functions. ... The representative methods were PHD and PSIPRED, and the overall precision of this generation was about 76% ...
Abstract. While the prediction of a native protein structure from sequence continues to remain a challenging problem, over the past decades computational methods have become quite successful in exploiting the mechanisms behind secondary structure formation. The great effort expended in this area has resulted in the development of a vast number ...
Initially, Burkhard Rost et al. [7] proposed the PHD method, which uses a contour alignment algorithm to automatically process the sequence contour consisting of amino acids and connect the sequence to the structure through a feedforward network, thus predicting the secondary structure of proteins. Subsequently, machine learning methods have ...
Example of a secondary structure prediction using PHD: ... Methods in Enzymology, 1996, 266, 513-525. version 2.2: Rob B. Russell & Andrei N. Lupas, 1999 _____ no coiled-coil above probability 0.5 _____ PHD: Profile fed neural network systems from HeiDelberg ~~~~~ Prediction of: secondary structure, by PHDsec solvent accessibility, by PHDacc ...
The PHD method by Rost & Sander (1993) uses a set of feed-forward neural networks trained by back-propagation (Rumelhart et al., 1986) to replace the "human expert" components of the Benner & Gerloff approach, and has since become the de facto standard secondary structure prediction method.
In recent years, deep neural networks have become the primary method for protein secondary structure prediction. Previous studies showed that deep neural networks had uplifted the accuracy of three-state secondary structure prediction to more than 80%. Favored deep learning methods, such as convolutional neural networks, recurrent neural ...
Secondary structure prediction methods are not often used alone, but are instead often used to pro-vide constraints for tertiary structure prediction methods or as part of fold recognition methods (e.g. Russelletal.,1996;Rost,1997). Early methods for secondary structure prediction were based on either simple stereochemical prin-
Because of its simple assumptions, the GOR method has conceptual advantage over other later developed methods such as PHD , PSIPRED , SPINE-X , and others. While these secondary structure prediction tools rely on machine learning and typically are black boxes in terms of the principles leading to their predictions, as we briefly review below ...
The prediction methods include Chou‐Fasman, Garnier, Osguthorpe and Robson (GOR), PHD, neural network (NN)‐based protein secondary structure prediction (PSIPRED), SPINE‐X, protein secondary structure prediction (PSSpred) and meta methods. The chapter assesses the performance of different methods using the Q 3 measure. It investigates the ...
The prediction methods include Chou-Fasman, Garnier, Osguthorpe and Robson (GOR), PHD, neural network (NN)-based protein secondary structure prediction (PSIPRED), SPINE-X, protein secondary structure prediction (PSSpred) and meta methods. The chapter assesses the performance of different methods using the Q3 measure.
Background The accuracy of protein secondary structure prediction has steadily improved over the past 30 years. Now many secondary structure prediction methods routinely achieve an accuracy (Q3) of about 75%. We believe this accuracy could be further improved by including structure (as opposed to sequence) database comparisons as part of the prediction process. Indeed, given the large size of ...
The first step is to select the secondary structure prediction methods (see below) you want to use. Then, the secondary structure consensus prediction program generates a secondary consensus. In this consensus the most present predicted conformational state is reported for each residue. Secondary structure prediction methods available are : DPM.
The first step in a PHD prediction is generating a multiple sequence alignment. ... Prediction of Secondary Structure at Better than 72% Accuracy PHDsec was the first secondary structure prediction method to surpass a level of 70% overall three-state per-residue accuracy. 6 The last test set with more than 300 unique protein chains and a total ...
years, secondary structure prediction is still an open problem, and few ad-vances in the eld have been made in recent times. In this thesis, the problem of secondary structure prediction is rstly ana-lyzed, identifying ve di erent information sources related to the biological essence of the problem, in order be exploited in a learning system. After
Although they differ in method, the aim of secondary structure prediction is to provide the location of alpha helices, and beta strands within a protein or protein family. Methods for single sequences. Secondary structure prediction has been around for almost a quarter of a century. The early methods suffered from a lack of data.
Protein structure prediction is one of the main research areas in the field of Bio-informatics. The importance of proteins in drug design attracts researchers for finding the accurate tertiary structure of the protein which is dependent on its secondary structure. In this paper, we focus on improving the accuracy of protein secondary structure prediction. To do so, a Multi-scale convolutional ...
This section of the paper discusses some of the popular methods used to predict protein secondary structure. PHD is a two-layered feed-forward neural network that uses evolutionary information to predict the secondary structure of water-soluble proteins. It was trained using 130 protein chains and increased prediction accuracy to 70.8% [17 ...
A total of 25 fungal isolates, categorized into 17 morphotypes, were obtained using the culture-dependent approach; Curvularia and Nigrospora emerged as the most common genera. Furthermore, the prediction of the ITS2 secondary structure supports the identification of species, highlighting a wide variety of fungal species present in H. binata.